Копнин Б.П.
ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России, Москва
3.3. р53 - многофункциональный опухолевый супрессор, чаще всего поражаемый в различных новообразованиях человека
3.3.1. Типы опухолей, ассоциированные с аномалиями р53
Наиболее универсальным молекулярным изменением в различных новообразованиях человека является инактивация функции белка р53. Более чем в половине всех опухолей человека (50-60% новообразований более чем 50 различных типов) обнаруживаются мутации гена р53. В отличие от других опухолевых супрессоров, для которых характерны мутации, прекращающие синтез белка (делеции, образование стоп-кодонов, сдвиг кодирующей рамки, нарушения сплайсинга мРНК), подавляющее большинство (более 90%) мутаций р53 представляет собой миссенс-мутации, приводящие к замене одной из аминокислот в белковой молекуле на другую. Еще одной особенностью мутаций р53 в опухолевых клетках является то, что они, в отличие от мутаций других опухолевых супрессоров, часто являются гетерозиготными, т.е. поражают только один из двух аллелей гена. (Причины этих различий будут раскрыты ниже, при рассмотрении структурной организации и функций р53 - см. раздел 3.3.2.)
Мутации обнаруживаются в разных участках молекулы р53, но чаще всего в его эволюционно консервативном ДНК-связывающем домене, причем с наибольшей частотой в кодонах 175, 245, 248, 249, 273 и 282 (так называемые горячие точки) - см. Рис. 2. Интересно, что спектр мутаций несколько меняется в зависимости от гистогенеза опухоли и/или этиологического фактора. Например, мутации в кодоне 175 не встречаются в опухолях легких, а замены в другой горячей точке - кодоне 273 - не обнаруживаются при бластном кризе хронического миелоидного лейкоза. В то же время для раков легкого характерны мутации в кодоне 145, очень редко встречающиеся в других новообразованиях, а мутации в кодоне 249 обнаруживаются преимущественно в гепатокарциномах, вызванных специфическим канцерогеном - афлатоксином В. Очевидно, отражением действия канцерогенов с разными механизмами мутагенного действия являются и отличия в характере мутаций р53 в разных опухолях. Так, при опухолях лёгких, печени и лимфомах замена аминокислотных остатков в большинстве случаев обусловлена трансверсиями (в ДНК пуриновый нуклеотид заменен на пиримидиновый или наоборот), а при карциномах кожи, лимфоме Беркитта, Т-клеточном лейкозе - транзициями (заменой пуринового основания на другое пуриновое или пиримидинового на другое пиримидиновое).
Герминальные (произошедшие в половой клетке и передающиеся по наследству) мутации в одном из аллелей гена р53 вызывают синдром Ли-Фраумени, заключающийся во врожденном предрасположении к развитию различных новообразований, в первую очередь сарком, рака молочной железы, лимфолейкозов. Нередко синдром Ли-Фраумени характеризуется возникновением первично-множественных опухолей. Примечательно, что у трансгенных мышей, несущих инактивирующие мутации в гене р53, наблюдается картина, очень напоминающая синдром Ли-Фраумени. Примерно у трети животных, у которых инактивирован один из двух аллелей р53, в течение 6-9 месяцев после рождения возникают новообразования, причем их спектр очень сходен с наблюдаемым при синдроме Ли-Фраумени. При этом в части этих опухолей, как и новообразований у пациентов с синдромом Ли-Фраумени, сохраняется экспрессия неповрежденного аллеля гена р53. При врожденной инактивации во всех клетках организма обоих аллелей гена р53 опухоли развиваются практически у всех животных. Примерно такая же картина наблюдается у трансгенных мышей, несущих дополнительный экзогенный аллель р53, кодирующий белок с миссенс-мутацией.
Важно подчеркнуть, что мутации - не единственный путь нарушения функции белка р53 в опухолевых клетках. Так, для 10-20% рака молочной железы, а также для нейробластом характерно нарушение транспорта р53 из цитоплазмы в ядро, где он проявляет свою функциональную активность. В части остеосарком наблюдается амплификация клеточного онкогена MDM2, продукт которого связывает и инактивирует белок р53 (см. раздел 3.3.3). При раке шейки матки, ассоциированном с вирусами папиллом человека, происходит связывание р53 с вирусным онкобелком Е6), что вызывает деградацию белка р53, и т.д.
3.3.2. Структурная организация и биохимические активности белка р53
Продукт гена р53 имеет мол. массу 53кДа и состоит из 392 аминокислотных остатков. Он образует тетрамерный комплекс, способный регулировать транскрипцию ряда генов, имеющих в своем составе специфические последовательности ДНК, так называемые р53-респонсивные элементы. В молекуле р53 картировано несколько функционально-значимых доменов, играющих важную роль в осуществлении или регуляции его активности (Рис. 2).
N-концевой участок (аминокислоты 1-42) представляет собой домен, ответственный за транскрипционную активацию генов-мишеней. Он обладает способностью связываться с компонентами базальных факторов транскрипции, в частности с субъединицами hTAFII31, hTAF70 комплекса TFIID РНК-полимеразы II, а также с транскрипционным кофактором p300/CBP. Кроме того, этот домен участвует в белок-белковых взаимодействиях, регулирующих стабильность молекулы р53. И, наконец, в нем расположено несколько остатков серина и треонина, фосфорилирование которых регулирует активность р53.
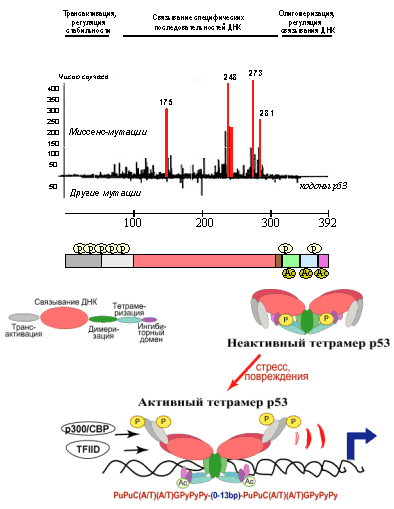
Рис. 2. Схематическое изображение функциональных доменов р53, предполагаемой модели приобретения белком транскрипционно активной конформации и частоты встречаемости в новообразованиях человека мутаций в разных участках молекулы р53.
Центральный домен р53 (аминокислоты 120-290) непосредственно узнает и связывает специфические последовательности ДНК регулируемых генов, так называемые р53-респонсивные элементы, состоящие из расположенных друг за другом последовательностей с обобщенной структурой типа PuPuC(A/T)(A/T)GPyPyPy (Pu - пурин, Py - пиримидин). Именно в этом ДНК-связывающем домене локализуется большинство точечных мутаций, обнаруживаемых в различных опухолях человека (Рис. 2).
Далее идут участки ответственные за ядерную локализацию (аминокислоты 305-323) и димеризацию/тетрамеризацию молекул р53 (аминокислоты 323-356). С-концевой участок р53 (аминокислоты 363-392) представляет собой так называемый ингибиторный домен. В немодифицированном состоянии он препятствует посадке ДНК-связывающего домена на специфическую последовательность регулируемого гена. Фосфорилирование и ацетилирование его определенных сайтов вызывают изменения конформации белковой молекулы и переход тетрамеров р53 из неактивного (латентного) состояния в активное. В результате ДНК-связывающие домены освобождаются от блокирующего влияния ингибиторных доменов и приобретают способность садиться на р53-респонсивные элементы. Таким образом, к респонсивным генам привлекаются базовые факторы транскрипции, связывающиеся с N-концевым участком р53, и стимулируется синтез РНК генов-мишеней.
Помимо повышения транскрипции генов, содержащих специфические респонсивные элементы, белок р53 обладает также и рядом других активностей. В частности, он способен подавлять транскрипцию многих других генов, например протоонкогенов BCL2, JUN и FOS, гена фибронектина и т.д. В основе такой транс-репрессии лежит несколько механизмов: связывание и секвестрация активированным р53 ряда базовых факторов транскрипции (p300/CBP, TBP, CBF); способность связывать и рекрутировать к определенным генам гистоновые деацетилазы (HDAC), ремоделирующие хроматин; и т.д. Кроме того, р53 связывается с белками, вовлеченными в репликацию или репарацию ДНК и, как следствие, модулирует эти процессы. Так, взаимодействуя с белком RP-A, он ингибирует его способность активировать ДНК-полимеразы a и d, результатом чего является подавление репликации ДНК. Связывая компоненты комплекса TFIIH (ERCC2, ERCC3 и др.), р53 активирует его функцию и стимулирует тем самым эксцизионную репарацию ДНК. Связывание р53 с белком Rad51 ведет к стимуляции рекомбинаций ДНК и повышению эффективности репарацию двунитевых разрывов ДНК. На участие р53 в репарации ДНК указывает также и его способность проявлять активность 3'-5'-экзонуклеазы и узнавать участки одноцепочечной ДНК и/или неспаренные основания.
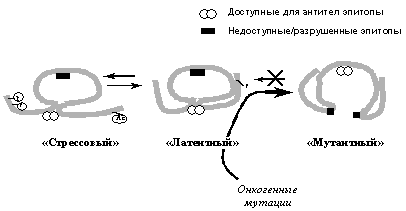
Рис. 3. Схематическое изображение различных конформационных состояний р53, распознаваемых специфическими антителами. Онкогенные мутации вызывают необратимый переход молекулы в денатурированное состояние, при котором открывается ранее недоступный эпитоп и, наоборот, исчезают некоторые ранее доступные эпитопы.
Характерные для опухолевых клеток миссенс-мутации приводят к резкому изменению конформации молекулы белка р53 (Рис. 3), что в значительной степени затрагивает все из вышеуказанных его активностей: происходит потеря или ослабление способности связывать и активировать гены с р53-респонсивными элементами, репрессировать другие специфические гены-мишени, ингибировать репликацию ДНК и стимулировать репарацию ДНК. Причем, так как р53 образует тетрамерные комплексы, мутации в одном аллеле гена р53 вызывают инактивацию и продукта второго, неповрежденного аллеля. Дело в том, что коэкспрессирующиеся нормальный и мутантный белок р53 образуют неактивные гетеромерные комплексы. Таким образом, мутантный белок ингибирует функции нормального белка р53 по доминантно-негативному механизму. По-видимому, именно эта особенность мутантных р53 в значительной мере ответственна за их онкогенный потенциал. В пользу этого свидетельствует тот факт, что введение в клетки короткого полипептида, соответствующего олигомеризационному домену р53, нарушает образование полноценных тетрамерных комплексов р53 и вызывает опухолевую трансформацию. Необходимо заметить, что помимо утраты нормальных функций р53, мутантные р53 с аминокислотными заменами в горячих точках (кодоны 175, 248 и др.) приобретают новые свойства, не свойственные белку р53 дикого типа (gain-of-function). Так, описано приобретение мутантными р53 способности активировать промоторы протоонкогенов MYC и ERB1, антиапоптотического гена BGL1 из семейства Bcl2, гена MDR1, детерминирующего множественную лекарственную устойчивость клеток и т.д. Предполагается, что это обусловлено способностью некоторых мутантных р53 связывать белки, в частности другие факторы транскрипции, с которыми нормальный р53 не взаимодействует, и модифицировать экспрессию генов, регулируемых этими транскрипционными факторами.
3.3.3. Физиологические функции р53 и их нарушения в неопластических клетках
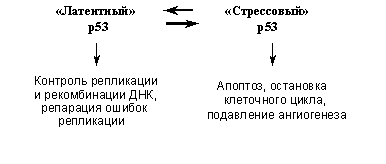
Молекулы белка р53 могут находиться в различных конформационных состояниях, в которых они обладают разными биохимическими активностями и выполняют разные физиологические функции. В обычных условиях р53 находится в так называемой латентной форме, в которой он обладает слабой транскрипционной активностью. Такой р53, однако, связывает белки репарационной машины (см. выше), проявляет активность 3'-5'-экзонуклеазы и стимулирует рекомбинацию и репарацию ДНК. При различных стрессах и внутриклеточных повреждениях происходят пост-трансляционные модификации, в частности фосфорилирование и ацетилирование определенных аминокислот молекулы р53, определяющие ее переход в так называемую стрессовую конформацию. Такой р53 значительно более стабилен (т.е. резко увеличивается его количество в клетке) и эффективно транс-активирует и/или транс-репрессирует специфические гены-мишени, следствием чего является индукция в аномальных клетках либо остановки клеточного цикла, либо апоптоза. Кроме того, активация р53 ведет к изменению экспрессии генов некоторых секретируемых факторов, в результате чего может изменяться размножение и миграция не только поврежденной, но и окружающих клеток. При этом, находясь в стрессовой конформации, р53 в значительной степени утрачивает активности, стимулирующие рекомбинацию и/или репарацию ДНК.
р53 дикого типа, в дополнение к латентной и стрессовой, может временно приобретать и так называемую мутантную конформацию, сходную с той, в которую молекула р53 необратимо переходит при онкогенных мутациях. Транзиторный переход р53 в мутантную конформацию происходит при воздействии определенных цитокинов и/или морфогенов (PDGF, тромбопоэтин, ретиноевая кислота и др.). Биологический смысл такого перехода пока неясен. Возможно, он заключается в полной инактивации рост-ингибирующих активностей р53 и/или изменении набора его генов-мишеней.
Таким образом, р53 играет важную охранную роль, являясь по образному выражению D.Lane, "стражем генома". Его повседневная функция заключается, по-видимому, в распознавании и исправлении ошибок, неизменно возникающих в ходе репликации ДНК. При массивных повреждениях ДНК, других внутриклеточных нарушениях или угрозе их возникновения происходит переключение функций р53 (Рис. 4): приобретая транскрипционные активности и изменяя экспрессию генов-мишеней, он вызывает либо остановку размножения аномальных клеток (временную, для устранения повреждений, или необратимую), либо их гибель (факторы, определяющие судьбу клетки при активации р53 будут рассмотрены ниже). В результате устраняется возможность накопления в организме генетически измененных клеток.
Механизмы активации р53 при стрессах и внутриклеточных повреждениях. Активация транскрипционных функций р53 наблюдается при самых разнообразных стрессах и внутриклеточных нарушениях: УФ- и g-облучении, присутствии в клетке разорванной ДНК, понижении внутриклеточного пула нуклеотидов, ингибировании ДНК- и РНК-полимераз, гиперэкспрессии онкогенов, вирусной инфекции, гипоксии, оксидативном стрессе, гипо- и гипертермии, различных нарушениях клеточной архитектуры (увеличении числа ядер, изменениях цитоскелета и адгезии) и т.д.
Ключевую роль в стабилизации белка р53 и повышении его транскрипционной активности играют изменения взаимодействия р53 с белком-ингибитором Mdm2, ген которого является потенциальным онкогеном. Белок Mdm2 связывается с N-концом молекулы р53 и, обладая активностями Е3 убиквитин-лигазы, стимулирует убиквитинизацию и, как результат, протеосомную деградацию белка р53. Поэтому, в норме уровень экспрессии р53 очень невелик, а время его жизни составляет всего около 30 минут. Кроме того, связываясь с N-концевым участком р53 в районе домена, взаимодействующего с базовыми факторами транскрипции, Mdm2 подавляет способность р53 транс-активировать гены-мишени.
A)
Б)
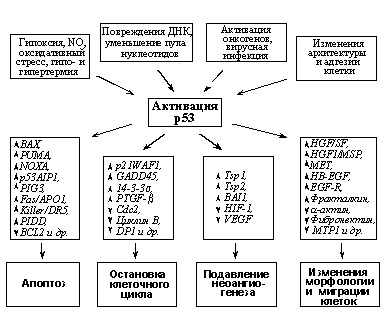
Рис. 4. Охранные функции р53.
А). Функции "латентной" и "стрессовой" форм р53.
Б) Факторы, вызывающие транскрипционную активацию р53, гены-мишени активированного р53 и вызываемые изменениями их экспрессии биологические эффекты.
При внутриклеточных нарушениях, в частности при повреждениях ДНК, происходит фосфорилирование р53 по сайтам (Ser15, Ser20, Ser33), расположенным в районе связывания с белком Mdm2. Такое фосфорилирование осуществляют специфические киназы (ATM, ATR, и их мишени - чекпойнткиназы CHK1, CHK2), которые активируются в ответ на разнообразные нарушения структуры ДНК (см. раздел 3.11.1). Кроме того, ATM фосфорилирует и белок Mdm2. В результате блокируется связывание р53 с Mdm2, что вызывает стабилизацию молекул р53 и повышение их транскрипционной активности. При некоторых других внутриклеточных изменениях, например при экпрессии в клетке активированных онкогенов RAS, также наблюдается нарушение взаимодействия р53 и Mdm2, но происходит оно из-за повышения экспрессии белка pARF, продукта альтернативной рамки считывания гена INK4a (см. раздел 3.4). Белок pARF обладает способностью связываться либо с N-концевым участком р53, либо с белком Mdm2, препятствуя, таким образом, их непосредственному взаимодействию друг с другом. Интересно, что ген MDM2 сам является транскрипционной мишенью активированного р53. В результате устанавливаются регуляторная петля, стимулирующая деградацию белка р53 после окончания действия факторов, вызывающих его фосфорилирование или связывание с белком pARF.
Важную роль в приобретении молекулами р53 конформации, способной транс-активировать гены-мишени, играют также модификации С-концевого участка, а именно ацетилирование его определенных аминокислотных остатков. При повреждениях ДНК и экспрессии активированного онкогена RAS эти события инициируются связыванием освобождающегося от Mdm2 N-концевого участка p53 с базальным фактором транскрипции р300/CBP, ацетилирующим сначала ингибиторный домен р53 по лизинам 373 и 382, а затем (после связывания р53 с респонсивными элементами) и белки хроматина в области генов-мишеней. Таким образом, последовательные пост-трансляционные модификации N-концевого и С-концевого участков р53 вызывают увеличение количества белка р53 в клетке, приобретение им способности связывать р53-респонсивные элементы и рекрутировать к генам мишеням базовые факторы транскрипции (компоненты комплекса TFIID РНК-полимеразы II и гистоновые ацетилазы p300/CBP, деконденсирующие хроматин), стимулируя тем самым транскрипцию их мРНК.
При некоторых стрессах, в частности при гипоксии, наблюдаются пост-трансляционные изменения р53, вызывающие его переход не к классической стрессовой конформации, а к ее варианту. Такой р53 не транс-активирует гены, содержащие р53-респонсивные элементы, но подавляет транскрипцию других генов-мишеней. Эта, так называемая репрессионная, форма также фосфорилирована по N-концу, но ее С-концевой участок не ацетилирован и связывает репрессионные комплексы Sin3/HDAC, вызывающие конденсацию хроматина генов-мишеней.
Гены-мишени р53 и их функции. В настоящее время, помимо Mdm2, обеспечивающего регуляцию самого р53 по принципу обратной связи (см. выше), идентифицировано более сотни генов, являющихся мишенями транскрипционных активностей р53. Они могут быть разделены на несколько групп, исходя из их физиологических функций (Рис. 4Б).
Первую группу составляют гены, продукты которых регулируют клеточный цикл. Важнейшим из них является белок p21Waf1/Cip1 - ингибитор циклинзависимых киназ из семейства Cip/Kip. Повышение его экспрессии вызывает остановку клеточного цикла в поздней фазе G1, что обусловливается связыванием им комплексов циклин Е/Сdk2, подавлением их способности фосфорилировать белки семейства рRb и освобождать транскрипционные факторы E2F (см. раздел 3.2.2). Дублирующим механизмом остановки перехода из G1 в S является подавление активированным р53 транскрипции гена DP1 - транскрипционного фактора, который связывается с E2F и образует активный комплекс, собственно и активирующий синтез продуктов, необходимых для входа в фазу S. Заслуживает также внимания способность р53 повышать экспрессию гена Siah1, продукт которого стимулирует деградацию b-катенина - транскрипционного фактора, активирующего транскрипцию гена циклина D и онкогена MYC (см. раздел 3.4.2), что вносит дополнительный вклад в индукцию остановки клеточного цикла в G1. Следует заметить, что р53 может подавлять активность Cdk2 не только в результате изменения своих транскрипционных функций, но и за счет белок-белковых взаимодействий, а именно непосредственного связывания циклина Н - компонента киназного комплекса CAK, осуществляющего активирующее фосфорилирование циклинзависимых киназ.
Идентифицирован также ряд генов-мишеней р53, продукты которых вызывают остановку в фазе G2 (задержка в ней наблюдается в случае, когда р53 активировался уже после того как клетка прошла G1-чекпойнт, или в клетках с инактивированным G1-чекпойнтом). Активированный р53 подавляет функцию комплекса циклин B/Cdc2, играющего ключевую роль в переходе из G2 в митоз, по нескольким механизмам. Во-первых, он транс-активирует ген 14-3-3-s, белковый продукт которого связывает и секвестрирует комплексы циклин B/Cdc2 в цитоплазме, не давая возможности им попасть в ядро, где они и должны проявлять свою активность. Во-вторых, он транс-активирует ген GADD45, белковый продукт которого обладает способностью связывать Сdc2, разрушая таким образом комплексы циклин B/Cdc2. В-третьих, р53 репрессирует транскрипцию генов циклина B и Cdc2, что уменьшает синтез их продуктов. Следует заметить, что, как и в случае остановки клеточного цикла в G1, задержка в G2 при повреждениях ДНК наблюдается и в клетках с инактивированным р53: она происходит в результате подавления функции фосфатаз Cdc25 (Cdc25A при остановке в G1 и Cdc25C при остановке в G2), активирующих соответствующие цикдинзависимые киназы. Однако в клетках с нарушенной функцией р53 происходит лишь кратковременная задержка в чекпойнтах, а активация р53 обеспечивает длительную остановку клеточного цикла, предотвращающую размножение вплоть до исправления дефекта.
Следующая группа р53-регулируемых генов кодирует белки, индуцирующие апоптоз. При этом р53 контролирует синтез компонентов обоих основных путей индукции апоптоза: и митохондриального, и стимулируемого "рецепторами смерти". Так, он регулирует активность белков семейства Bcl2, репрессируя ген анти-апоптотического белка Bcl2 и активируя гены про-апоптотических белков Bax, Puma и Noxa. Повышение проницаемости митохондриальной мембраны для цитохрома С и белка AIF достигается и транс-активацией гена p53AIP1 (его продукт локализуется в митохондриальной мембране и уменьшает мембранный потенциал) и гена PIG3 (кодирует оксиредуктазу, которая вовлечена в образование радикалов кислорода, повреждающих мембраны митохондрий и стимулирующих выброс их содержимого). Стимуляция апоптоза, запускаемого рецепторами смерти достигается транс-активацией генов двух из таких рецепторов - Fas и Killer/DR5 (рецептор для TRAIL). Еще одной такой мишенью является ген недавно идентифицированного белка Pidd, который содержит домен смерти и при гиперэкспрессии индуцирует апоптоз. Выявлены и другие р53-респонсивные гены (IGF-BP3, PAG608, p85, циклин G), продукты которых стимулирует апоптоз, однако механизмы их апоптогенного действия пока исследованы довольно плохо. Кроме того, р53 может индуцировать апоптоз и через другие механизмы, несвязанные с его способностью изменять экспрессию генов-мишеней. Так, мутантные р53, утратившие транскрипционную активность (в результате делеции С-концевого участка или мутаций в кодонах 22-23), сохраняют тем не менее способность индуцировать апоптоз в некоторых (но не во всех) типах клеток. Предполагается, что этот эффект обусловлен белок-белковыми взаимодействиями р53. Таким образом, при повышении функциональной активности р53 может происходить активация сразу многих путей индукции апоптоза, что, по-видимому, обеспечивает надежность его реализации.
Так как р53 контролирует активность генов, продукты которых способны вызвать как остановку клеточного цикла в различных его фазах, так и апоптоз, возникает вопрос, отчего зависит выбор судьбы клетки при активации р53. Выяснилось, что он определяется множеством факторов: гистогенетическим типом клеток (например, в нормальных фибробластах, как правило, наблюдается остановка клеточного цикла, тогда как в лимфоцитах - апоптоз), степенью активации р53 (с увеличением уровня его экспрессии повышается вероятность апоптоза), функциональной активностью сигнального пути pRb-E2F (в фибробластах c инактивированным pRb или гиперэкспрессированным E2F, наблюдается не остановка в G1, а апоптоз) и т.д. Недавно обнаружено, что еще одним фактором, определяющим выбор между остановкой клеточного цикла и апоптозом, является характер модификации самих молекул р53 и/или их белок-белковых взаимодействий. Так р53, фосфорилированный по Ser15/20 и ацетилированный по С-концу, способен транс-активировать ген p21Waf1/Cip1 и вызывать остановку в G1, тогда как дополнительное фосфорилирование по Ser46 придает ему способность транс-активировать наряду с геном p21Waf1/Cip1 и ген белка р53AIP1, а в этом случае уже наблюдается апоптоз. Причем вероятность фосфорилирования Ser46 повышается с увеличением интенсивности повреждений ДНК. Кроме того, способность р53 избирательно транс-активировать проапоптотические гены (BAX и др.) увеличивается при его связывании с белками семейства ASPP (ASPP1 и ASPP2), потеря экспрессии которых характерна для значительной части случаев рака молочной железы.
Третьей большой группой генов-мишеней р53 являются гены, продукты которых регулируют морфологию и/или миграцию клеток (Рис. 4Б). Так, р53 транс-активирует гены обоих представителей семейства рассеивающих (Scatter) факторов - HGF/SF и HGF1/MSP, а также ген одного из членов семейства эпидермальных факторов роста HB-EGF (гепарин-связывающий EGF) (продукты всех этих генов являются одновременно и митогенами, и мотогенами). При этом, р53 транс-активирует также гены рецепторов этих факторов - HGF/SF-R (Met) и EGF-R. р53-респонсивными также являются гены хемокина фракталкина, гладкомышечного a-актина, коллагенов II1 и Vl1 типов, ингибитора плазминогена PAI-1. С другой стороны р53 репрессирует гены фибронектина и металлопротеиназы I типа. Физиологическое значение такой регуляции пока не установлено. Возможно, оно заключается, по крайней мере частично, в привлечении к клеткам с активированным р53 окружающих клеток определенных типов для ремоделирования/восстановления структуры ткани в месте возможного апоптоза. Не исключено участие такой регуляции и в процессах морфогенеза.
В особую группу генов-мишеней р53 можно выделить гены, контролирующие ангиогенез. Ключевую роль в неоангиогенезе играет VEGF (Vascular Endothelial Growth Factor; стимулирует размножение и миграцию эндотелицитов), экспрессия которого повыщается при гипоксии или активации некоторых онкогенов. р53 репрессирует транскрипцию как гена VEGF, так и гена HIF-1 (Hypoxia Inducing Factor 1) - транскрипционного фактора, обеспечивающего повышение экспрессии VEGF и его рецепторов в ответ на уменьшение содержания кислорода (кроме этого HIF-1 изменяет экспрессию генов, контролирующих транспорт глюкозы и гликолиз, что обеспечивает адаптацию клеток к условиям гипоксии). Одновременно р53 может транс-активировать гены белков, ингибирующих ангиогенез - тромбоспондинов (Tsp)-1, -2 (связывая специфические рецепторы на поверхности эндотелиоцитов, они вызывают в них апоптоз) и BAI-1. Таким образом клетки с активированным р53 хуже переносят недостаток кислорода, перестают секретировать VEGF и начинают секретировать ингибиторы ангиогенеза, что препятствует образованию новых сосудов. Данные функции являются, по-видимому, еще одной составляющей опухоль-супрессирующего действия р53, так как они предотвращает адаптацию к гипоксии и прорастание сосудов в центр опухоли.
Выявлено еще несколько десятков генов-мишеней р53. Среди них следует отметить ген каталитической субъединицы теломеразы (TERT), который репрессируется р53 (таким образом, р53 участвует, по-видимому, и в обеспечении репликативного старения клеток. По-видимому, р53 принимает участие и в процессах созревания клеток, так как некоторые из транс-активируемых им генов кодируют белки репертуара той или иной дифференцировки (мышечная креатинкиназа и др.).
Последствия нарушений функции р53. Характерные для опухолевых клеток аномалии р53 отменяют или ослабляют все важнейшие функции опухолевого супрессора р53. Делеция обоих аллелей гена р53, т.е. его полная инактивация, вызывает ослабление G1- и G2-чекпойнтов клеточного цикла, подавление индукции апоптоза, уменьшение эффективности репарации ДНК, более эффективную адаптацию к гипоксии и стимуляцию неоангиогенеза, ослабление контроля за длиной теломер, ингибирование дифференцировки и другие характерные свойства неопластической клетки. Особо следует отметить возникновение в клетках с инактивированным р53 сильной генетической нестабильности, являющейся мотором дальнейшей опухолевой прогрессии. Потеря функциональной активности р53 значительно увеличивает темп появления размножающихся клеток с самыми разными генетическими аномалиями - измененным числом и перестройками хромосом, генными мутациями, амплификацией отдельных участков генома.
Сходные последствия наблюдаются и при самых частых в новообразованиях человека аномалиях р53 - миссенс-мутациях, ведущих к синтезу неактивного белка, обладающего доминантно-негативным эффектом в отношении продукта неповрежденного аллеля. Следует заметить, однако, что степень проявления доминантно негативного эффекта мутантного р53 варьирует в зависимости от конкретной аминокислотной замены, типа клеток и ингибируемой функции р53 дикого типа. Поэтому нередко селективное преимущество получают клоны клеток, в которых в результате дополнительных генетических событий происходит делеция или мутация и второго аллеля гена р53. В то же время, опухолевые клетки, как правило, сохраняют экспрессию хотя бы одного мутантного аллеля гена р53. Очевидно, это связано с тем, что появляющиеся в результате миссенс-мутаций новые активности мутантных р53 вносят дополнительный вклад в повышение онкогенного потенциала клетки. Так, приобретая способность транс-активировать онкоген MYC, мутантный р53 вызывает, очевидно, более сильные нарушения регуляции клеточного цикла, чем те, которые наблюдаются при делециях гена р53. Новые активности мутантных белков р53 ответственны также за дополнительное ослабление индукции апоптоза и развитие устойчивости к действию химиопрепаратов. В основе подавления апоптоза лежит несколько механизмов: транс-активация мутантным р53 гена антиапоптотического белка Bag1 (член семейства Bcl2), способность мутантных р53 связывать и инактивировать гомолог р53, белок р73 (см. следующий раздел) и т.д. Возникновение устойчивости к определенным противоопухолевым цитостатикам может быть связано также со способностью некоторых мутантных р53 повышать транскрипцию гена MDR1 (Multi-Drug Resistance 1 - см. раздел XII.2.1.1) и гена дУТФ-азы, продукт которого блокирует действие 5-фторурацила и ряда других антиметаболитов. Следует заметить, что характерные для опухолей человека мутации дифференциально влияют на возникновение вышеуказанных активностей. Так, замены в кодонах 175 и 248 придают способность активировать ген дУТФ-азы, тогда как мутации в кодоне 273 не вызывают приобретения такого свойства. Поэтому, использование мутаций р53 в качестве критерия для предсказания чувствительности к той или иной химиотерапии может быть основано лишь на точной идентификации аминокислотных замен, а не на гистохимической детекции "нормального" или "мутантного" р53 с помощью конформационно-специфических антител.
Таким образом, мутации и другие изменения активности р53 вызывают одновременное появление целого набора характерных свойств неопластической клетки, таких как понижение чувствительности к различным рост-супрессирующим сигналам (в том числе генерируемым перманентной экспрессией активированных онкогенов), иммортализация, повышение способности выживать в неблагоприятных условиях, генетическая нестабильность, стимуляция неоангиогенеза, блокирование клеточной дифференцировки и т.д. Это, очевидно, и является объяснением такой частой встречаемости мутаций р53 в самых разных новообразованиях - они позволяют за один шаг преодолеть сразу несколько этапов опухолевой прогрессии. Причем, мутации р53 могут являться как инициальным событием (синдром Ли-Фраумени) или детерминировать начальные этапы канцерогенеза, так и возникать и отбираться уже в ходе роста опухоли, обеспечивая приобретение новых агрессивных свойств и устойчивости к терапии.
3.3.4. Гомологи р53: р63 и р73
Недавно были обнаружены два гена, р63 и р73, продукты которых имеют достаточно высокую степень гомологии с белком р53 в участках транс-активационного, ДНК-связывающего и олигомеризационного доменов, но значительно отличающиеся от него по C-концу. Однако, в отличие от гена р53, кодирующего по существу один белковый продукт, подвергающийся, правда, самым разным пост-трансляционным модификациям, гены р63 и р73 продуцируют по несколько белков. Дело в том, что мРНК каждого из них может синтезироваться с двух промоторов и далее подвергаться альтернативному сплайсингу. В результате образуется по 6 изоформ белков р63 и р73, обладающие (ТА-формы) или не обладающие (ΔDN-формы) транс-активационным доменом. Причем формы, содержащие транс-активационный домен способны активировать р53-респонсивные гены и вызывать, в частности, апоптоз. Кроме того, ТА-формы р63 и р73 могут транс-активировать и ряд других генов, содержащих близкие по структуре респонсивные элементы, которые не являются, тем не менее, мишенями р53. В частности, они транс-активируют гены белков Jag1 и Jag2 - лигандов для рецепторов Notch, активация которых играет ключевую роль в выборе судьбы клетки при выборе направления ее дифференцировки.
Существует еще ряд отличий между р53 и его гомологами. Так, если р53 экспрессируется в клетках практически всех тканей, то его гомологи экспрессируются только в некоторых типах клеток. Например, р63 преимущественно экспрессируется в эмбриональных клетках, а также стволовых и недифференцированных эпителиальных клетках взрослого организма, причем в виде транскрипционно-неактивных ΔDN-форм. Предполагается, что экспрессия ΔDN-форм р63 обеспечивает недифференцированное состояние клетки. При нокауте гена р63 у мышей наблюдается пренатальная или постнатальная гибель эмбрионов вследствие полного отсутствия кожи и других эпителильных тканей (предполагается, что это связано с преждевременной дифференцировкой стволовых клеток эпителия и исчерпанием их запаса к моменту рождения животных). Далее, если р53 активируется в ответ на самые разные стрессы, то его гомологи только на некоторые из них, причем за счет совершенно других механизмов. Наконец, если р53 выполняет функции опухолевого супрессора и для новообразований характерна его инактивация, то его гомологи не имеют таких функций. Об этом свидетельствует две группы фактов. Во-первых, у мышей гомозиготный нокаут гена р73 не вызывает повышения частоты возникновения опухолей (нокаут гена р63 не совместим с жизнью эмбрионов - см. выше). Во-вторых, в опухолях человека не выявляется потеря экспрессии или мутации генов р63 и р73. Наоборот, в них нередко отмечается повышение экспрессии этих белков, причем, как правило, транскрипционно-неактивных ΔDN-форм (т.е. р63 и р73 являются скорее протоонкогенами). В связи с этим приобретает популярность гипотеза, согласно которой ΔDN-формы белков р63 и р73 функционируют как природные ингибиторы р53, подавляющие его функцию по доминантно-негативному механизму. Действительно, их транскрипционно-неактивные тетрамеры могут конкурировать с р53 за места посадки на ДНК генов-мишеней, а мономеры/димеры - секвестрировать р53 путем связывания его молекул и образования неактивных комплексов. Это не исключает, однако, что при нарушениях функции р53 происходит активация транскрипционной функции его гомологов (показано, что р53 репрессирует транскрипцию гена р73), которая может частично компенсировать утрату функции р53 и обеспечивать, например, нормальное развитие мышей с гомозиготным нокаутом гена р53. Эти предположения, как и другие аспекты биологических функций гомологов р53, требуют дальнейшего исследования.