Копнин Б.П.
ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России, Москва
1.2. Механизмы возникновения характерных свойств неопластической клетки
Приобретение совокупности перечисленных свойств связано с нарушениями в сигнальных путях, контролирующих реакцию клетки на экзогенные и эндогенные регулирующие факторы. Ключевую роль в их возникновении играют изменения регуляции клеточного цикла, апоптоза, миграции и некоторых других базовых процессов жизнедеятельности клетки.
1.2.1. Нарушения регуляции клеточного цикла
Нарушения регуляции клеточного цикла лежат в основе таких важнейших свойств неопластической клетки как самодостаточность в пролиферативных сигналах (пониженная потребность во внешних сигналах для инициации и поддержания пролиферации) и нечувствительность к рост-супрессирующим сигналам. Кроме того, они в значительной степени определяют генетическую нестабильность и нарушения дифференцировки клетки.
В процессе подготовки клетки к делению и образования из нее двух новых клеток наблюдается несколько фаз - G1 фаза, в которой идет подготовка к синтезу ДНК; S фаза - период репликации ДНК; G2 фаза, в которой идет подготовка к митозу, и, наконец, собственно митоз - процесс разделения клетки на две новые. Образовавшиеся дочерние клетки могут сразу войти в новый митотический цикл или временно выйти из него в G0 фазу - стадию покоя. "Мотором" клеточного цикла является активация последовательно сменяющих друг друга циклинзависимых киназ (Рис. 4). Каждая из циклинзависимых киназ представляет собой холоферментный комплекс, состоящий из собственно каталитической субъединицы (Cdk) и регуляторной субъединицы - циклина. Связывание с циклином увеличивает киназную активность Cdk и, кроме того, определяет их локализацию и субстратную специфичность. Уровень экспрессии каждого из циклинов и, в меньшей степени, Cdk, направленно изменяется в определенные фазы клеточного цикла. Так, выход клетки из стадии покоя и вход в фазу G1 определяется образованием комплексов циклинов D (D1-D3) с Сdk4 или Cdk6 (в зависимости от типа клеток). Переход из G1 в S связан с образованием комплексов циклина E с Сdk2, и т.д. Вход в митоз, например, обусловлен образованием комплексов Сdk1 (другое и более распространенное название - Cdc2) с циклином В
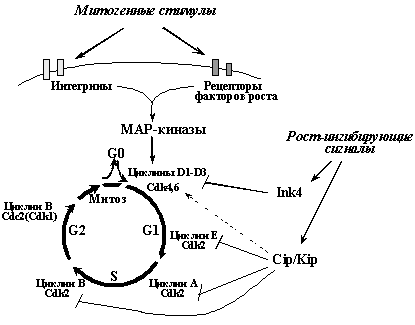
Рис.4. Общие принципы регуляции клеточного цикла в нормальной клетке. Вход в цикл и продвижение по нему определяется последовательной активацией различных циклинзависимых киназ (Cdk). Повышение их активности инициируется сигналами от рецепторов ростовых факторов и интегринов (рецепторов белков внеклеточного матрикса), вызывающими сложный каскад событий, приводящий к активация группы МАР-киназ - ключевых регуляторов Cdk. Центральную роль в негативной регуляции размножения клеток клеток играют ингибиторы циклин-зависимых киназ (CKI) - белки семейств Ink4 и Cip/Kip. Для опухолевых клеток характерны изменения, ведущие к перманентной стимуляции активности циклинзависимых киназ и подавлению функции их ингибиторов.
Кроме связывания с циклинами, активность Сdk регулируется изменениями фосфорилирования их определенных аминокислотных остатков (за такую регуляцию ответственны фосфатазы Cdc25 и протеинкиназы CAK, Wee1) и связыванием с так называемыми CKI - ингибиторами Cdk. CKI - группа белков, состоящая из двух семейств. Представители семейства Cip/Kip (p21WAF1/CIP1, p27KIP1 и p57KIP2) ингибируют различные комплексы Cdk2, ответственные за вход и продвижение по S фазе. В меньшей степени они подавляют активность комплексов циклин B/Cdc2, ответственных за вход в митоз. С другой стороны, они не ингибируют, а даже активируют комплексы циклин D/Cdk4(6), оперирующие в ранней G1 фазе. Члены семейства Ink4 (p16INK4a, p15INK4b, p18INK4c, p19INK4d) непосредственно взаимодействуют с Cdk4(6). При этом, связывая Cdk4(6), находящиеся в составе активных комплексов с циклином D и белками Cip/Kip, они вытесняют белки Cip/Kip, направляя их на связывание с Cdk2. Поэтому повышение активности белков Ink4, в частности опухолевого супрессора p16Ink4a , вызывает как прямое ингибирование активности комплексов циклин D/Cdk4(6), так и непрямое блокирование комплексов циклин E/Cdk2 и циклин А/Cdk2.
Выход покоящейся клетки из фазы G0 и вступление ее в митотический цикл инициируется различными внешними стимулами, в первую очередь различными секретируемыми цитокинами, принадлежащими к группе факторов роста. Кроме этого, для деления большинства типов нормальных клеток необходимо также и взамодействие специфических рецепторов клетки - интегринов - с определенными белками внеклеточного матрикса (фибронектином, коллагеном и др.). Связывание рецепторов со своими лигандами - ростовыми факторами и белками внеклеточного матрикса - индуцирует аутофосфорилирование внутриклеточных доменов рецепторов и дальнейшее их взаимодействие со многими сигнальными белками. Следствием этого является стимуляция пересекающихся сигнальных путей и активация так называемых МАР (Mitogen Activated Protein) киназных каскадов. Конечные продукты этих каскадов - серин-треониновые киназы ERK1/2, JNK и р38 - транслоцируются из цитоплазмы в ядро, где они фосфорилируют множество субстратов, что, в конце концов, и приводит к активации циклинзависимых киназ, иницирующих вход в S фазу.
Действие большинства рост-ингибирующих факторов (цитокинов типа TGF-b, контактного торможения при установлении межклеточных контактов, повреждений ДНК и других внутриклеточных нарушений) основано на активации ингибиторов циклинзависимых киназ (CKI) семейств Ink4 и Cip/Kip, что приводит к остановке клеточного цикла в так называемых чекпойнтах (checkpoints, сверочных точках). В зависимости от типа рост-ингибирующего воздействия и молекул, вовлеченных в его распознавание, наблюдается остановка клеточного цикла в G1, S, G2 фазах или в митозе. При этом, задержка в G2 связана как с активацией CKI семейства Cip/Kip, так и с повышением активности ряда других молекул, ингибирующих функцию комплексов циклин В/Cdc2, а остановка в митозе - с изменениями активности молекул, контролирующих конденсацию хромосом и разделение сестринских хроматид (Рис. 5).
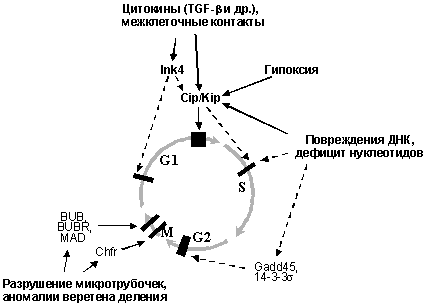
Рис.5. Активация чекпойнтов клеточного цикла (обозначены черными прямоугольниками) при различных рост-ингибирующих воздействиях.
Для опухолевых клеток характерны генетические изменения, вызывающие, с одной стороны, перманентную стимуляцию сигнальных путей, активирующих Сdk4(6) и Cdk2, а с другой стороны - нарушения в путях передачи сигналов, опосредующих активацию чекпойнтов в ответ на рост-ингибирующие сигналы. Первый тип изменений возникает в основном в результате активирующих мутаций так называемых протоонкогенов - нормальных компонентов путей передачи различных митогенных сигналов (см. раздел II.2) и приводит к самодостаточности в пролиферативных сигналах, т.е. способности неопластической клетки постоянно генерировать внутри себя сигналы к размножению, в норме исходящие от внешних стимулов. Инактивация чекпойнтов, обеспечивающая нечувствительность к рост-ингибирующим сигналам и генетическую нестабильность, чаще всего обусловлена дисфункцией т.н. опухолевых супрессоров, к которым относятся и некоторые из CKI.
С изменениями регуляции клеточного цикла связаны и нарушения дифференцировки неопластических клеток. Реализация большинства дифференцировочных программ требует выхода клетки из митотического цикла в стадию покоя (G0). Перманентная стимуляция размножения клеток и нечувствительность к действию рост-ингибирующих цитокинов, являющихся одновременно индукторами дифференцировки (таких как TGF-b) нередко приводят к блокированию или извращению процессов дифференцировки.
1.2.2. Изменения морфологии и движения клеток
Верхние этажи сигнальных путей, активируемых связыванием рецепторов ростовых факторов и интегринов со своими лигандами, ответственны за стимуляцию не только размножения, но и движения клеток. Поэтому многие из цитокинов являются одновременно и митогенами, и мотогенами (например, EGF, HGF/SF, PDGF и др.). Активация G-белков семейства Ras и фосфатидилинозит-3-киназы (PI3K), находящихся на пересечении сигнальных путей от многих рецепторов, ведет к повышению активности как МАР-киназ - ключевых регуляторов клеточного цикла, так и малых ГТФ-аз семейства Rho (Rho, Rac, Cdc42), играющих центральную роль в реорганизации цитоскелета и регуляции движения клеток.
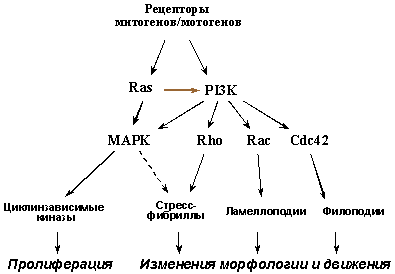
Рис. 6. Верхние этажи сигнальных путей, активируемых рецепторами ростовых факторов, контролируют как размножение, так и движение клеток.
Для опухолевых клеток характерны генетические изменения (активирующие мутации рецепторных тирозинкиназ, протоонкогенов семейства RAS и др. - см. раздел II.2), вызывающие конститутивную стимуляцию такой сигнализации (Рис. 6). В результате возникают клетки с повышенным пролиферативным потенциалом, характерными изменениями морфологии и увеличенной способностью к миграции, а, следовательно, и к инвазивному росту и метастазированию. Клетка с такими изменениями, возникшими, например, в результате активации протоонкогенов RAS, приобретает также и способность самостоятельно продуцировать и секретировать ряд митогенов/мотогенов, включая VEGF (Vascular Endotelial Growth Factor), стимулирующий размножение и направленную миграцию окружающих эндотелиоцитов, т.е. неоангиогенез.
1.2.3. Отсутствие репликативного старения (иммортализация)
Известно, что при культивировании in vitro человеческие фибробласты после 60-70 делений перестают размножаться и останавливаются, преимущественно в G1 фазе клеточного цикла. Очень редко, примерно в одной клетке из нескольких миллионов, происходят спонтанные мутации, отменяющие остановку клеточного цикла. Однако потомки этой клетки делятся еще порядка 10 раз и снова останавливают свое размножение, а затем постепенно гибнут. Первая остановка клеточного цикла получила название "ранний кризис", или стадия М1, а вторая остановка и гибель клеток стали определяться термином "кризис", или стадия М2 репликативного старения. В пользу того, что остановка и гибель клеток связана именно с числом предшествующих клеточных делений, а не просто с астрономическим временем, проведенным вне организма, в условиях in vitro, свидетельствует две группы фактов. Во-первых, в культурах фибробластов, полученных от эмбрионов или от молодых людей, кризис наступает позже, чем в культурах фибробластов, полученных от пожилых людей. Во-вторых, если до наступления стадии М1 фибробласты не пересаживать, они образуют плотную культуру и, в результате контактного торможения, остановятся в G1. В этом состоянии они могут находиться довольно долго, по крайней мере 2-3 месяца, а затем, после пересадки, снова начнут размножаться до тех пор, пока не пройдут свои 60-80 делений. В это время их аналоги, не останавливавшие свое размножение, уже давно погибли. Таким образом, астрономическое время пребывания в культуре может быть увеличено, а неизменным остается число клеточных делений до наступления кризиса. Если же какие-то клетки избегают кризиса (спонтанная частота такого события настолько низка, что ее даже трудно зарегистрировать, но определенные манипуляции с геномом или экспрессия некоторых клеточных и вирусных онкогенов могут значительно увеличить вероятность этого события), то клетки могут уже размножаться бесконечно долго. Это определяется термином иммортализация клеток, т.е. приобретение бессмертия.
В основе счетно-ограничительтного механизма, детерминирующего репликативное старение клеток, лежит прогрессивное укорочение теломер (концевых участков хромосом) по мере деления клеток. ДНК теломер, представляющая собой более тысячи повторов гексануклеотида TTAGGG, в силу известной проблемы репликации концов линейной ДНК, синтезируется не полностью, и при каждом акте репликации, т.е. после каждого клеточного деления, теломеры укорачиваются. Согласно теломерной гипотезе прогрессивное укорочение теломер приводит к тому, что они достигают какой-то критической минимальной длины, когда сенсорные системы начинают распознавать их как аномальные структуры ДНК и индуцировать остановку клеточного цикла, подобно тому, как это происходит при ДНК-повреждающих воздействиях. Именно это, как сейчас предполагается, и вызывает фазу М1 репликативного старения (ранний кризис). При нарушениях в сигнальных системах, детерминирующих остановку клеточного цикла, клетки с укороченными теломерами будут продолжать делиться, пока теломеры практически не исчезнут и перестанут выполнять свои функции, т.е. предотвращать рекомбинации и слипание хромосом. Тогда хромосомы потеряют свою целостность и наступит так называемая генетическая катастрофа, или стадия М2 репликативного старения (Рис.7).
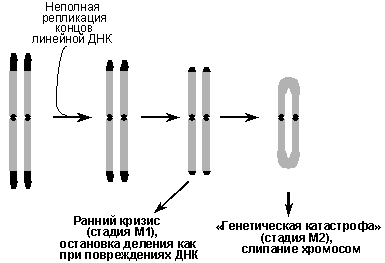
Рис. 7. Теломерный механизм репликативного старения клеток человека
Отсутствие в опухолевых клетках человека репликативного старения (иммортализация) связано с включением специального механизма. В его основе лежит способность специфического фермента теломеразы достраивать недореплицированные теломерные повторы и поддерживать таким образом их постоянную длину. Теломераза состоит из нескольких субъединиц, включая РНК-матрицу и TERT (Telomerase Reverse Transcriptase), представляющую собой обратную транскриптазу, синтезирующую ДНК повторов гексануклеотида TTAGGG с РНК-матрицы. Предполагается существование и других, т.н. ALT (альтернативных) механизмов поддержания длины теломер, основанных на гомологичной рекомбинации ДНК теломер. Включение экспрессии TERT, каталитической субъединицы теломеразы, индуцируется изменением экспрессии определенных онкогенов (см. раздел II.2) или опухолевых супрессоров. Так, оно может быть вызвано активацией онкогена Myc и инактивацией опухолевого супрессора р53. Кроме этого, существенный вклад в иммортализацию неопластических клеток вносят нарушениями работы охранных механизмов, осуществляющих остановку клеточного цикла при нарушения структуры ДНК, в частности при исчезновении теломерных повторов (см . раздел 1.2.1).
При обычных условиях культивировании in vitro в некоторых типах клеток человека, например в эпителиоцитах, наблюдается остановка клеточного цикла через 10-15 делений - т.н. фаза М0 репликативного старения. Такая остановка не связана, по-видимому, с изменением длины теломер, а обусловливается активацией CKIs, в частности p16INK4a, в ответ на какие-то внутриклеточные изменения, вызываемые неадекватностью условий in vitro. Так фаза М0 в кератиноцитах или эпителиоцитах молочной железы может быть отменена при культивировании их на подложке фибробластов в среде без сыворотки, но с достаточным содержанием набора определенных факторов роста. Сходная ситуация наблюдается в культурах фибробластов и других клеток грызунов, которые входят в кризис через 10-15 делений in vitro несмотря на очень большую длину теломер и высокую активность теломеразы в подавляющем большинстве клеток. Как и в случае эпителиальных клеток человека, кризис в клетках грызунов может быть предотвращен подбором адекватных условий культивирования (определенный для каждого типа клеток внеклеточный матрикс и набор цитокинов), в связи с чем он получил название "культуральный шок" (culture shock). Таким образом, совсем недавно выяснилось, что у клеток грызунов, в отличие от клеток человека, нет механизма подсчета числа делений (т.е. они как бы изначально иммортализованы благодаря конститутивной активности теломеразы, поддерживающей довольно большую длину теломер), а их репликативное старение представляет собой артефакт, связанный с неадекватностью условий in vitro. Описанные ранее события, вызывающие иммортализацию фибробластов грызунов (инактивация опухолевых супрессоров р53, pARF) или отмену фазы М0 в эпителиоцитах человека (инактивация опухолевых супрессоров p16Ink4a, pRb) предотвращают активацию чекпойнтов клеточного цикла в ответ на внутриклеточные изменения, накапливающиеся при неправильном культивировании клеток. Кроме того, они могут отменять индукцию терминальной дифференцировки факторами сыворотки крупного рогатого скота, которая является причиной артефактного репликативного старения некоторых типов клеток, например крысиных олигодендроцитов и шванновских клеток.
1.2.4. Изменения регуляции апоптоза
Апоптоз вызывается различными сигналами, как физиологическими - экспрессией специальных киллерных цитокинов, изменениями гормонального статуса (цикличное ремоделирование эндометрия матки, возрастная инволюция тимуса и др.), так и нефизиологическими - различными внутриклеточными повреждениями или неблагоприятными условиями - нехваткой факторов роста, повреждениями ДНК, гипоксией и т.д. В регуляции апоптоза выделяют два основных этапа: фазу индукции (принятия решения) и фазу экзекуции (исполнения приговора). Последняя осуществляется путем активации каспаз - семейства цистеиновых протеиназ, расщепляющих свои субстраты по остаткам аспартатовой кислоты. Расщепление каспазами 3, 6, 7 (так называемые эффекторные, или казнящие каспазы) ряда ключевых субстратов, в частности ингибиторов нуклеаз, ламинов - ядерных цитоскелетных белков и т.д., приводит к фрагментации ДНК и деструкции клетки. Каспазы присутствуют в цитоплазме в виде проэнзимов и активируются до полностью функциональных протеаз путем расщепления проэнзима на большую и малую субъединицы и дальнейшего отщепления от них N-концевых доменов. Затем субъединицы собираются в активные олигомеры. Расщепление прокаспаз могут осуществлять различные протеазы, в том числе и другие каспазы.
Существует два принципиально разных сигнальных пути, приводящих к активации эффекторных каспаз 3, 6, 7 (Рис. 8). Один из них инициируется связыванием специфических киллерных лигандов (Fas-лиганд, TNF-a и др.) со своими рецепторами, так называемыми рецепторами смерти, что вызывает рекрутирование к ним адаптерных белков и прокаспаз, в частности прокаспазы 8 (подробнее - см. раздел Недоспасова). Агрегация молекул прокаспазы 8 достаточна, чтобы инициировать их аутопроцессирование (расщепление) и образование активных форм каспазы 8, которая, в свою очередь, процессирует до активных форм эффекторные каспазы.
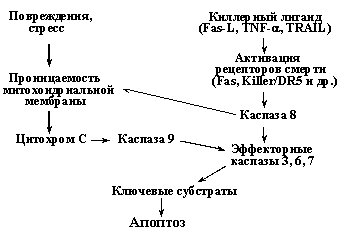
Рис. 8. Два основных пути активации каспаз и индукции апоптоза.
При альтернативном пути индукции апоптоза ключевую роль играют митохондрии, поэтому его называют митохондриальным путем. Он иницируется главным образом различными повреждающими воздействиями, вызывающими увеличение проницаемости митохондриальной мембраны и выход в цитоплазму ряда митохондриальных белков, в частности цитохрома С, который связывается с белком APAF-1 и стимулирует образование его олигомеров. Это, в свою очередь вызывает рекрутирование на образовавшийся комплекс молекул прокаспазы-9, их агрегацию, аутопроцессирование и формирование активного комплекса каспазы-9. На следующем этапе происходит рекрутирование на этот комплекс молекул прокаспазы-3 и их процессирование до активных форм, которые расщепляют ключевые мишени и вызывают апоптоз. Следует заметить, что повреждения и стрессы приводят к выходу из митохондрий не только цитохрома С, но и ряда других молекул, в частности протеазы AIF (Apoptosis Inducing Factor). AIF также стимулирует апоптоз, вызывая расщепление ряда ядерных белков каспазонезависимым путем. Таким образом, митохондриальный путь индукции апоптоза включает несколько независимых механизмов (Рис. 9). При этом, существует взаиморегуляция сигнальных путей, активируемые рецепторами смерти и повреждающими воздействиями (Рис. 8). Так, активация каспазы 8, вызванная активацией рецепторов смерти, не только напрямую активирует эффекторные каспазы, но и индуцирует увеличение проницаемости митохондриальной мембраны и активацию регуляторной каспазы 9. С другой стороны, при повреждениях и стрессах может наблюдаться повышение экспрессии рецепторов смерти - Fas и Killer/DR5. Таким образом, проапоптотические сигналы амплифицируются, что позволяет надежнее достигать необходимого эффекта.
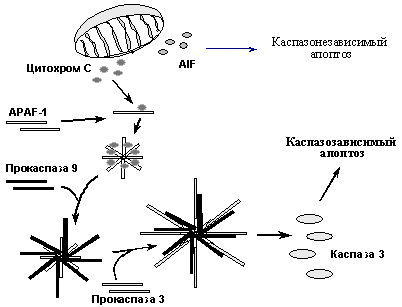
Рис. 9. Механизмы митохондриального пути индукции апоптоза.
Ключевую роль в регуляции проницаемость митохондриальной мембраны для цитохрома С и AIF играют белки семейства Bcl2, подразделяющиеся на несколько подсемейств и обладающие либо проапоптотическими, либо антиапоптотическими активностями. Предполагается, что антиапоптотические белки (Bcl-2, Bcl-X, и др.), локализуясь в мембранах митохондрий, закрывают каналы, через которые осуществляется выброс цитохрома С и AIF. Проапоптотические молекулы (Bax, Bad и др.) при апоптогенных сигналах перемещаются из цитоплазмы в митохондриальные мембраны, где взаимодействуя с интегральным белком наружной митохондриальной мембраны VDAC, стимулируют открытие канала, через который, в частности секретируется цитохром С. Кроме того, белки подсемейства Bax образуют гетеромерные комлексы с белками Bcl2, Bcl-x, что, возможно, открывает закрытые до этого каналы.
Для опухолевых клеток характерны генетические изменения, ведущие к ослаблению обоих путей индукции апоптоза. Так, в них закономерно обнаруживаются:
потеря экспрессии на поверхности клетки рецептора смерти Fas;
нарушения проведения апоптогенного сигнала к митохондриям (например, при инактивации опухолевых супрессоров р53 и PTEN);
ингибирование проницаемости митохондриальной мембраны для цитохрома С и AIF, вследствие изменений экспрессии белков семейства Bcl2;
блокирование активации эффекторных каспаз (например, при потере экспрессии белка APAF-1 в результате метилирования его гена);
резкое уменьшению времени жизни каспаз ввиду их связывания с белками IAP (Inhibitors of Apoptosis), экспрессия которых повышается вследствие активации протоонкогенов Ras, PKB/Akt (см. раздел II.2) или инактивации опухолевого супрессора PTEN
1.2.5. Генетическая нестабильность
Генетическая нестабильность - это увеличение вероятности возникновения и закрепления в ряду клеточных поколений разнообразных изменений генома. Важность приобретения этого признака для образования злокачественной опухоли связана с тем, что именно генетическая нестабильность, вместе с постоянно идущим отбором, обеспечивают накопление в одной клетке сразу нескольких мутаций в онкогенах, опухолевых супрессорах и других генах, придающих клетке совокупность необходимых для образования опухоли свойств. Генетическая нестабильность популяций опухолевых клеток складывается из 4 основных типов нарушений:
а) уменьшения точности воспроизведения генетической информации, а именно понижения точности репликации ДНК и сегрегации хромосом во время митоза;
б) нарушений в системах репарации повреждений ДНК или ошибок, возникших при ее репликации;
в) ослабления функции чекпойнтов клеточного цикла, активируемых в ответ на повреждения структуры ДНК или веретена деления в митотической клетке, в результате чего клетка, несмотря на разрывы ДНК или изменения числа хромосом, продолжает делиться и умножать число аномальных потомков;
д) ослабления индукции апоптоза, вследствие чего делящиеся клетки с генетическими нарушениями не погибают, а выживают.
Совокупность перечисленных нарушений обеспечивает повышенную частоту возникновения различных генетических изменений и их закрепление в ряду клеточных поколений.
Понижение точности репликации ДНК в неопластических клетках связано с повышением синтеза и активности в них так называемых низкоточных полимераз, в частности ДНК-полимеразы-b, которая в норме используется лишь для быстрой репарации массивных повреждений ДНК (основную роль в воспроизводстве ДНК играет высокоточная ДНК-полимераза-d). Повышение содержания низкоточных ДНК-полимераз b, e и др. может быть вызвано экспрессией ряда онкогенов, например BCR/ABL, RAS. Нарушения правильной сегрегации хромоосом во время митоза могут происходить вследствие изменения числа и структуры центросом или центров организации микротрубочек: в опухолевых клетках нередко обнаруживается больше двух центросом, что ведет к многополярным митозам и возникновению анеуплоидных вариантов с неправильным числом хромосом. К увеличению числа центросом в клетке приводит активация онкогена RAS и инактивация опухолевых супрессоров р53, APC или BRCA1. Остальные компоненты генетической нестабильности неопластических клеток - нарушения работы систем репарации ДНК, инактивация чекпойнтов клеточного цикла и ослабление индукции апоптоза - рассмотрены в других разделах.
В последние годы выяснилось, что повышенная изменчивость популяций опухолевых клеток связана не только с резким учащением появления истинных генетических изменений (генных мутаций, рекомбинаций, анеуплоидии и др.), но и со значительным увеличением вероятности возникновения в неопластических клетках т.н. эпигенетических изменений. В основе таких изменений лежит ремоделирование структуры хроматина, обусловленное изменениями метилирования цитозинов ДНК и/или ацетилирования гистонов. В результате происходит подавление экспрессии одних генов и/или повышение экспрессии других генов, причем из-за характерных для опухолевых клеток нарушений процессов метилирования ДНК одновременно может изменяться транскрипция нескольких сотен генов