Копнин Б.П.
НИИ канцерогенеза,
Российский Онкологический научный центр, РАМН,
115478, Москва,
Каширское ш., 24;
факс: (095)324-1739;
электронная почта: kopnin@imb.ac.ru
4. Онкогены, опухолевые супрессоры и нарушения морфогенетических реакций клетки
Ярким отличительным свойством неопластических клеток является "асоциальный" тип их поведения, связанный в первую очередь с нарушениями нормальных морфогенетических реакций - потерей контактного торможения размножения, приобретением способности к пролиферации независимо от прикрепления к субстрату, изменениями адгезионных взаимодействий, формы и подвижности клеток и т.д. Именно эти нарушения вместе с некоторыми другими свойствами, в частности способностью секретировать протеолитические энзимы и ангиогенные факторы, предопределяют инвазивный характер роста (проникновение в окружающие здоровые ткани), а впоследствии и метастазирование (образование вторичных очагов опухолевого роста) [118]. Первостепенную роль в возникновении указанных выше нарушений морфогенетических реакций играют изменения функции протоонкогенов и/или опухолевых супрессоров (рис.7, 8).
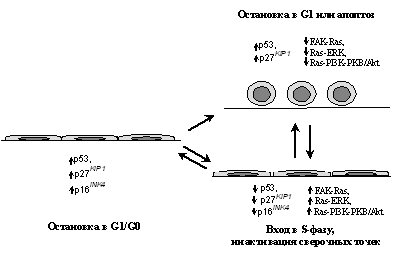
Рис. 7. Изменения активности супрессорных белков и контролируемых протоонкогенами сигнальных путей, определяющие зависимость клеток от прикрепления к субстрату и контактное торможение размножения (объяснения в тексте)
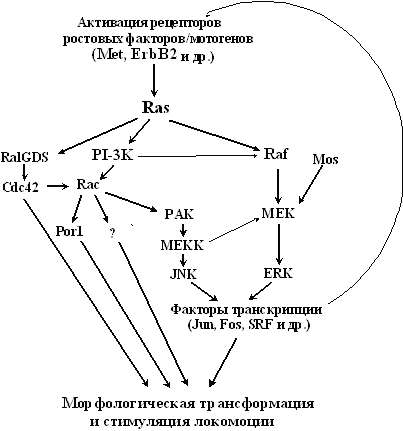
Рис. 8. Регулируемые онкогенами сигнальные пути, ответственные за изменения морфологии и локомоторный фенотип неопластических клеток (объяснения в тексте)
Присущее нормальным клеткам контактное торможение размножения (прекращение пролиферации при установлении контактов с окружающими клетками) связывают в первую очередь с повышением экспрессии опухолевых супрессоров p16INK4a и p27KIP1 [37,119,120], что обусловливает недофосфорилирование pRb и блокирование входа в S-фазу [17] (см. раздел 1). Пути передачи сигнала от плазматической мембраны к ингибиторам циклинзависимых киназ пока неясны. Показано только, что повышение в эпителиальных клетках экспрессии Е-кадхерина, вызванное трансдукцией его гена, ведет к накоплению p27KIP1 и остановке клеточного роста [121]. Недавно появились данные о существовании в эпителиальных клетках еще одного пути блокирования клеточного цикла в ответ на установление межклеточных контактов. Продемонстрировано, что образование эпителиального пласта вызывает накопление р53, тогда как мутации Е-кадхерина и/или разобщение межклеточных контактов, наоборот, вызывают дестабизацию р53 и, как следствие, прекращение ингибирующего воздействия p21WAF1 на комплексы циклин - Cdk. В связи с этим можно думать, что онкогенный потенциал мутаций Е-кадхерина, ответственных за развитие наследственных форм рака желудка и многих других новообразований [122], по крайней мере частично обусловлен изменениями регуляции клеточного цикла, апоптоза и контроля генетической стабильности [123].
Наряду с инактивацией ряда опухолевых супрессоров (Е-кадхерин, р53, p27KIP1, pRb), вызванной мутациями или связыванием с вирусными онкобелками (pRb - c E1A, E7, T-SV40; p53 - c E1B, E6, T-SV40; p27KIP1 - c E1A и т.д. [7,10,124]), к потере контактного торможения размножения может приводить и гиперфункция протоонкогенов, модифицирующих активность сигнального пути Cdk-pRb-E2F. В частности, она может быть вызвана повышением экспрессии Myc или активацией протоонкогена Ras (первый вызывает деградацию p27KIP1 и трансактивацию Cdc25a, второй - деградацию p27KIP1 и повышение экспрессии циклина D1, см. раздел 1).
Независимость от прикрепления к внеклеточному матриксу (anchorage independence). Чтобы выживать и пролиферировать, нормальные клетки большинства типов должны быть прикреплены к внеклеточному матриксу. Два основных фактора лежат в основе этого явления: неспособность ростовых факторов активировать в неприкрепленных клетках комплексы циклин E - Cdk2, ответственные за вход в S-фазу [125], и индукция апоптоза во многих типах клеток при отсутствии адгезионных взаимодействий (этот тип апоптоза имеет специальное название "аноикис") [126]. И подавление пролиферации, и индукция апоптоза в неприкрепленных клетках могут быть связаны с активацией р53, вызываемой откреплением клеток от субстрата и отсутствием сигналов от рецепторов интегринов [58,127]. За подавление входа в S-фазу кроме активации сигнального пути р53-p21WAF1, по всей видимости, ответственна и аккумуляция p27KIP1, также закономерно наблюдающаяся при отсутствии контактов клеток с матриксом [127,128]. Однако помимо запуска механизмов негативного контроля пролиферации (блокирование входа в S-фазу и индукция апоптоза) в ответ на открепление клеток от матрикса существуют и независимые механизмы позитивной регуляции выживаемости и размножения клеток, инициируемые связыванием интегринов с белками внеклеточного матрикса и последующей активацией нерецепторной тирозинкиназы FAK (Focal Adhesion Kinase - ключевого участника передачи сигналов от интегриновых рецепторов, физически взаимодействующего с цитоплазматическим доменом b-субъединицы интегрина [118,126]). Во-первых, связывание интегринов с матриксом и активация FAK необходимы для прохождения митогенного сигнала от рецепторов ростовых факторов (EGF, PDGF) до конечных МАР киназ - ERK1/2. (В неприкрепленной клетке сигналы от этих ростовых факторов блокируются на промежуточной МАР киназе MEK1, которая в силу каких-то неизвестных пока причин не фосфорилирует свои мишени - ERK1/2 [64].) Во-вторых, активируя Ras через адаптерный белок Shc, интегриновые рецепторы не только стимулируют митогенный сигнал, но и подавляют аноикис (апоптоз), по всей видимости, за счет активации сигнального пути Ras-PI3K-PKB/Akt [126] (cм. раздел 2).
Если исходить из факта существования нескольких механизмов, определяющих зависимость жизнеспособности и/или размножения клеток от их связывания с матриксом, становится понятным, что для возникновения характерной для опухолевых клеток независимости от адгезионных взаимодействий необходимо, по-видимому, несколько событий, которые, с одной стороны, позволяли ли бы преодолевать супрессорные эффекты р53 (мутации/делеции этого гена, гиперэкспрессия онкогена MDM2 и др.) и/или p27KIP1 (мутации/делеции; гиперэкспрессия онкогенов RAS и MYC, приводящая к деградации данного белка, и др.), а с другой, - обходить прерывание митогенного сигнала на уровне МЕК1 киназы (например, за счет активации белков Src или Myc, вызывающих активацию комплексов циклин E - Cdk2) и блокировать аноикис по сигнальному пути Ras-PI3K-PKB/Akt.
Изменения формы и подвижности клеток. Изменение формы (морфологии) - характерное свойство опухолевых клеток, позволяющее при микроскопическом исследовании диагносцировать злокачественную трансформацию. В основе морфологических нарушений лежат связанные между собой изменения цитоскелета, адгезионных взаимодействий клеток друг с другом и с внеклеточным матриксом. Вкратце они выражаются в нарушении формирования фокальных контактов и в ухудшении прикрепления клеток к матриксу, дезорганизации системы актиновых микрофиламентов. Это приводит к изменениям активности псевдоподий и характера перемещения клеток. В целом наблюдаемая картина напоминает изменения, возникающие в нормальных клетках при действии мотогенных цитокинов - факторов, стимулирующих передвижение клеток. Однако так называемый локомоторный фенотип в неопластических клетках, как правило, сильно утрирован, что позволяет отличать по морфологии опухолевую клетку от движущейся нормальной клетки.
Молекулярные механизмы, определяющие возникновение локомоторного фенотипа, как в нормальных клетках при мотогенных стимулах, так и при неопластической трансформации, пока далеки от ясности. Выявлены лишь некоторые ключевые узлы в пересекающихся цепях передачи сигналов, ответственных за возникновение этих изменений. Как известно, многие цитокины (например, HGF/SF, EGF, FGF, PDGF, IGF-1 и др.) являются одновременно и митогенами, и мотогенами [118]. Так, HGF/SF (Hepatocyte Growth Factor/Scatter Factor) является сильным митогеном для гепатоцитов, но мотогеном для различных эпителиальных клеток, в частности клеток молочной железы, эндотелиоцитов и др. Мотогенный эффект HGF/SF зависит от вызываемой им стимуляции Ras-Raf-MAPК сигнальных путей: об этом свидетельствует его отмена при ингибирования функции МЕК1 и блокировании передачи сигнала к ERK1/2 [129-131]. В то же время одного только повышения активности Raf, по-видимому, недостаточно для разборки в эпителиальных клетках Е-кадхериновых межклеточных контактов и стимуляции локомоции клеток - необходима одновременная активация и PI3K, которая при действии HGF/SF может индуцироваться как по Ras-зависимому, так и по Ras-независимым сигнальным путям [130,132]. Эффекторы PI3K, ответственные за реализацию мотогенного эффекта, неизвестны. Показано только, что ни PKB/Akt, ни Rac сами по себе его не обеспечивают [130], хотя базальный уровень активности Rac, по-видимому, необходим для осуществления перестроек цитоскелета и разъединения межклеточных контактов, требующихся для локомоции [132]. Показано также, что одним из важных эффектов, вызываемых HGF/SF в клетках лейомиосаркомы, является понижение экспрессии гена фибронектина, что может изменять адгезионные взаимодействия с матриксом и локомоцию [133]. Мутации гена рецептора HGF/SF (протоонкогена Met), приводящие к перманентной стимуляции его тирозинкиназной активности, являются онкогенными: они способны вызывать морфологическую трансформацию в культивируемых in vitro клетках [134] и ответственны за развитие наследственного папиллярного рака почки, а также некоторых других заболеваний человека (табл. 1). Молекулярные основы мотогенных эффектов других цитокинов изучены еще хуже, но предполагается, что, как и в случае с HGF/SF, они связаны со стимуляцией Ras-PI3K и Ras-MAPК сигнальных путей [135-139] .
Конститутивная экспрессия активированных онкогенов Ras вызывает и в фибробластах, и в эпителиальных клетках резкое повышение локомоторной активности и стойкие морфологические изменения, характерные для неопластических клеток [140,141] (заметим, однако, что эти изменения хорошо проявляются только в клетках с аномалиями р53- и/или p16INK4a-регулируемых сигнальных путей; в остальных случаях в ответ на перманентную гиперэкспрессию Ras может индуцироваться либо апоптоз, либо остановка в G1, уменьшающая степень выраженности Ras-индуцированных морфологических изменений [117,142]. Предполагается, что несколько эффекторов Ras, в первую очередь Raf и GTPазы семейства Rho (Rac, Cdc42, Rho), ответственны за проявление морфологической трансформации и за стимуляцию локомоторной активности неопластических клеток [20,143] (рис. 8). Как и при мотогенных эффектах цитокинов, ключевую роль здесь, по-видимому, играет активация двух сигнальных каскадов: Ras-PI3K-Rac и Ras-Raf-ERK [20,143-145]. Для трансформации фибробластов, очевидно, достаточно конститутивной активации любого из этих путей. Действительно, трансдукция активированного Rac1, как и активированных Raf, Mos (подобно Raf стимулирует MEK1), или самой МЕК1 может вызывать морфологическую трансформацию фибробластов грызунов [146-150], а блокирование в Ras-трансформированных клетках функции либо MEK1, либо Rac, хотя и приводит к частичной реверсии морфологических изменений, но не предотвращает трансформацию [148,151]. В то же время в эпителиальных клетках, хорошо трансформирующихся онкогеном Ras, активации Raf-ERK-каскадов недостаточно, чтобы вызвать морфологическую трансформацию [149]. Экспрессия активированного Rac придает эпителиальнысм клеткам некоторые признаки трансформированного фенотипа (в первую очередь образование ламеллоподий и раффлинг мембран [143,152]), однако сильная морфологическая трансформация достигается, по-видимому, только при совместной активации Rac-зависимых и Raf-ERK сигнальных путей [144,153].
О молекулярных событиях, ответственных за морфологическую трансформацию при активации Rac и Raf-ERK сигнальных путей, известно очень мало. Стимуляция MAP киназ ERK1/2 ведет, как известно, к активации ряда транскрипционных факторов, в частности Elk1, Fos, SRF. Заметим, что одним из важнейших последствий гиперфункции Rac является стимуляция другого МАР киназного каскада (рис. 8), конечный продукт которого - JNK - вызывает активацию транскрипционных факторов Jun и ATF2. Таким образом, и Raf-ERK-, и Rac-JNK-сигнальные пути регулируют активность транскрипционных комплексов AP-1 (состоит из гомодимеров Jun/Jun или гетеродимеров Jun/Fos), функция которых представляется весьма важной для индукции морфологических изменений. Об этом свидетельствуют как способность самого онкогена Jun вызывать трансформацию клеток [154,155], так и реверсия трансформированного фенотипа при подавлении функции Jun [156,157]. Не исключено, впрочем, что трансформирующий потенциал Jun (AP-1) связан не с прямым действием на мишени, регулирующие адгезивные взаимодействия, организацию цитоскелета и локомоцию, а с образованием аутокринной петли, обусловленной стимуляцией продукции ростовых факторов/мотогенов (EGF и др.), которые в свою очередь активируют Ras и его эффекторы, в частности Rac и другие GTPазы семейства Rho (Cdc42, RhoA), ответственные за изменения морфологии [155,157] (рис. 8). Так предполагается, что Rac обеспечивает образование ламеллоподий и раффлинга, Cdc42 - вызывает образование филоподий, а RhoA участвует в формировании фокальных контактов и стресс-фибрилл [143,152].
Существующие модели рассматривают образование ламеллоподий как следствие Rac-индуцированного повышения активности белка Por1 [126,152], возникновение стресс-фибрилл - как последствие активации миозинфосфатазы под действием Rho-киназы, а образование фокальных контактов - как результат связывания с актином цитоскелетных белков профилина и гельзолина, стимулируемого фосфатидилинозитолбифосфатом - одной из мишеней Rho [126,152]. Пока неясно, однако, обеспечиваются ли все эти реакции передачей сигналов исключительно между цитоплазматическими белками или изменение активности множества транскрипционных факторов также вносит свой вклад в эти процессы.
Таким образом, главенствующую роль в морфологической трансформации и приобретении локомоторного фенотипа играют, очевидно, протоонкогены, изменения активности которых приводят к активации белков семейства Ras и/или его эффекторов - PI3K, Raf и, возможно, RalGDS. По-видимому, только совокупность вызываемых ими изменений в регуляции активности псевдоподий, сборки/разборки цитоскелета и фокальных контактов при одновременном изменении активности большого набора транскрипционных факторов обеспечивает в конце концов приобретение клетками так называемого "полностью трансформированного" фенотипа, определяющего агрессивный характер роста. Следует, однако, учитывать, что для проявления этих изменений необходима инактивация опухолевых супрессоров (p53 p19ARF и/или p16INK4a), предохраняющих организм от появления в нем клонов клеток с конститутивно активированными Ras-MAP-киназными сигнальными путями. Кроме того, выраженность морфологической трансформации и способности к локомоции уменьшаются при повышении экспрессии некоторых других опухолевых супрессоров - Е-кадхерина [118] и pRb [158]. Более того, два других важнейших признака неопластической клетки - ослабление контактного торможения размножения и приобретение независимости от субстрата - зависят в первую очередь от инактивации определенных опухолевых супрессоров (р53, p27KIP, Rb, Е-кадхерина). Поэтому кажется очевидным, что для возникновения характерных для опухолевых клеток изменений морфогенетических реакций требуется несколько генетических событий (мутаций), затрагивающих и опухолевые супрессоры, и протоонкогены.