Копнин Б.П.
НИИ канцерогенеза,
Российский Онкологический научный центр, РАМН,
115478, Москва,
Каширское ш., 24;
факс: (095)324-1739;
электронная почта: kopnin@imb.ac.ru
2. Онкогены и опухолевые супрессоры в регуляции апоптоза
Другой важнейшей точкой приложения активностей онкогенов и опухолевых супрессоров является регуляция апоптоза (программированной гибели клеток). Апоптоз, как известно, вызывается различными сигналами: связыванием с рецепторами специфических киллерных лигандов, нехваткой факторов роста/выживания, повреждениями ДНК и разрушениями цитоскелета, гипоксией и другими неблагоприятными условиями (см. обзоры [49-52]). В регуляции апоптоза выделяют два основных этапа: фазу индукции (принятия решения) и фазу экзекуции (исполнения приговора). Последняя осуществляется путем активации каспаз - семейства цистеиновых протеиназ, расщепляющих свои субстраты по остаткам аспартатовой кислоты. Расщепление каспазами 3, 6, 7 (так называемые "эффекторные" или "казнящие" каспазы) ряда ключевых субстратов, в частности DFF45/ICAD - ингибитора нуклеазы DFF40/CAD (осуществляется каспазой 3), ламинов - ядерных цитоскелетных белков (осуществляется каспазой 6) и т.д., приводит к фрагментации ДНК и деструкции клетки [52]. Каспазы присутствуют в цитоплазме в виде проэнзимов и активируются до полностью функциональных протеаз путем расщепления проэнзима на большую и малую субъединицы и дальнейшего отщепления от них N-концевых доменов. Затем субъединицы собираются в тетрамер с двумя активными центрами [49,52]. Расщепление прокаспаз могут осуществлять различные протеазы, в том числе и другие каспазы.
Предполагается, что существует по меньшей мере два принципиально разных сигнальных пути, приводящих к активации каспаз 3, 6, 7 [49,52] (рис. 5). Один из них инициируется связыванием специфических киллерных молекул (Fas-лиганд, TNFa и др.) со своими рецепторами, что вызывает рекрутирование адаптерных белков и прокаспаз, в частности прокаспазы 8. Агрегация молекул прокаспазы 8 достаточна, чтобы инициировать их аутопроцессирование (расщепление) и образование активных форм каспазы 8, которая, в свою очередь, процессирует "казнящие" каспазы. При альтернативном механизме расщепление каспаз 3, 6, 7 осуществляется каспазой 9, активация которой инициируется выходом из митохондрий протеазы AIF (Apoptosis Inducing Factor) и/или цитохрома с, стимулирующего связывание прокаспаз 9 с белком Apaf1 (гомолог белка CED-4 у C. elegans) и, как следствие, образование агрегатов прокаспаз 9 и аутопроцессирование их до активных форм. Проницаемость митохондриальной мембраны для AIF и цитохрома с регулируется белками семейства Bcl2. Это семейство структурно сходных белков включает более двух десятков членов, в том числе продукты протоонкогенов bcl2 и bcl-x, обладающие способностью блокировать апоптоз, и опухолевый супрессор Bax, наоборот, индуцирующий апоптоз [53-55]. Предполагается, что антиапоптогенные молекулы Bcl2 и Bcl-x, локализуясь в мембранах митохондрий, закрывают каналы, через которые осуществляется выброс цитохрома с и/или AIF. Bax, находящийся в норме в определенных компартментах цитоплазмы, при апоптогенных сигналах перемещается в митохондриальные мембраны, где он, взаимодействуя с интегральным белком наружной митохондриальной мембраны VDAC, стимулирует открытие канала, через который секретируется цитохром с. Кроме того, Bax образует гетеромерные комлексы с белками Bcl2, Bcl-x, что, возможно, открывает закрытые до этого каналы [54,55]. Другие проапоптотические белки семейства Bcl2 (Bak, Bad, Bid и т.д.), по-видимому, обладают сходным действием [53,55].
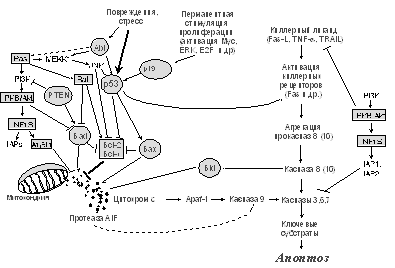
Рис. 5. Участие онкогенов и опухолевых супрессоров в регуляции апоптоза (объяснения в тексте)
Если Bcl2, Bcl-x и Bax непосредственно контролируют выброс из митохондрий апоптогенных молекул, то ряд других протоонкогенов и опухолевых супрессоров регулируют активность этих и других белков семейства Bcl2 (рис. 5). Одним из наиболее мощных таких регуляторов является опухолевый супрессор р53. Активируясь в ответ на самые разные неблагоприятные воздействия (повреждения ДНК, гипоксия, потеря контактов клетки с субстратом, перманентная нерегулируемая стимуляция митогенного сигнала и многие другие [13,14,56-58], p53 осуществляет на транскрипционном уровне одновременно и активацию гена bax, и репрессию гена bcl2 [57,59]. Кроме того, р53 повышает экспрессию ряда генов PIG, продукты которых вызывают оксидативный стресс и, как следствие, нарушения проницаемости митохондриальной и ядерной мембран [60], а также трансактивирует некоторые киллерные рецепторы, в частности Fas и KILLER/DR5 [57,61,62]. Таким образом, активация р53 дает мощный апоптогенный сигнал, в реализации которого задействованы различные механизмы индукции "казнящих" каспаз. Важно подчеркнуть, что р53-зависимый апоптоз элиминирует из организма не только поврежденные клетки, но и клетки, в которых наблюдается нерегулируемая стимуляция пролиферации, вызываемая, например, конститутивной активацией онкогена MYC и/или транскрипционного фактора E2F. Стабилизация р53 при активации онкогенов связана с индуцируемым E2F повышением транскрипции гена p19ARF, продукт которого препятствует Mdm2-зависимой деградации р53 [13,14]. Естественно, что инактивирующие мутации р53 или p19ARF, нарушающие работу этого защитного механизма, будут резко увеличивать вероятность появления постоянно пролиферирующих клеточных клонов, а следовательно, и вероятность последующего развития из них злокачественных опухолей.
Интересно, что конститутивная экспрессия онкогенов Ras инициирует одновременно и апоптогенные и антиапоптогенные сигналы. Первые обусловлены активацией сигнального пути Ras-Raf-МАРК-E2F-p19ARF-p53 [63]. Вторые связаны как со способностью одного из эффекторов Ras - белка Raf - прямо фосфорилировать и инактивировать проапоптотический белок Bad (член семейства Bcl2), так и с действием другого эффектора Ras - PI3K (фосфоинозитол-3-киназы) [21,51,63,64]. Антиапоптотические эффекты PI3K обусловлены ее способностью активировать серин-треониновую протеинкиназу PKB/Akt (впервые была идентифицирована как онкоген ретровируса AKT8, вызывающего Т-клеточные лимфомы у мышей AKR), которая блокирует апоптоз несколькими путями [65] (см. рис. 5). Во-первых, она, как и Raf, обладает способностью фосфорилировать и инактивировать белок Bad. Кроме того, супрессируя функцию белка DAF-16 - транскрипционного фактора семейства Forkhead - PKB/Akt может ингибировать продукцию киллерных молекул, в частности Fas-лиганда. И, наконец, недавно обнаружено, что PKB/Akt активирует функцию транскрипционных факторов семейства Rel/NF-kB [65] (гомологи вирусного онкобелка v-Rel; амплификация и перестройки их генов характерны для многих новообразований человека [66]), которые, в свою очередь, ингибируют апоптоз несколькими путями (рис. 5). В частности, он трансактивирует ген, кодирующий белок A1/Bfl1 - член семейства белков Bcl2, ингибирующий выброс цитохрома С и/или AIF [67]. Наряду с этим, NF-kB увеличивает экспрессию ингибиторов апоптоза IAP1 и IAP2, членов семейства белков IAP (Inhibitors of Apoptosis), блокирующих функцию каспаз 3, 6, 7, 8, 9 [66]. В связи с изложенным становится понятной одна из защитных функций опухолевого супрессора PTEN (его инактивация закономерно обнаруживается в глиомах, раках молочной железы и простаты, а врожденные мутации ведут к развитию синдрома множественных гамартом [68], табл. 2): белок PTEN, обладающий активностями тирозиновой фосфатазы, подавляет антиапоптогенные эффекты сигнала PI3K-PKB/Akt [69].
Для неопластических клеток характерны нарушения функции и других опухолевых супрессоров, осуществляющих позитивную регуляцию апоптоза. Так, развитие хронического миелоидного лейкоза обусловливается хромосомной транслокацией t(9;22), в результате которой образуется химерный ген BCR/ABL. Такая перестройка вызывает одновременно два важных последствия: а) резкое увеличение тирозинкиназной активности белка Abl, что ведет к стимуляции митогенного и антиапоптотического сигналов, опосредуемых Ras-регулируемыми сигнальными путями [70,71], а также увеличением синтеза интегринов, обеспечивающим лучшее прикрепление к внеклеточному матриксу [72], и б) инактивацию апоптогенных активностей Abl [73-75], обусловленных, по-видимому, его участием в позитивной регуляции JNK (другое название SAPK, Stress Activated Protein Kinase), которая обладает способностью подавлять активность Bcl2 и, возможно, активировать р53 (рис. 5). Ряд данных указывают также на то, что белок Abl может непосредственно связываться с р53, модифицируя его проапоптотическую функцию [57,65,75].
Результатом хромосомной транслокации t(15;17), наблюдающейся в подавляющем большинстве случаев острого промиелоцитарного лейкоза, является соединение гена рецептора ретиноевой кислоты (RAR-a) c геном опухолевого супрессора PML [3,12,76], продукт которого образует в ядре специфические матрикс-ассоциированные тельца. Предполагается, что химерный белок PML/RAR-a инактивирует по доминантно-негативному механизму апоптогенную функцию нормального белка PML, образуя с ним гетеродимеры. Механизмы индукции апоптоза при гиперэкспрессии PML пока не совсем ясны. Получены данные об его участии в активации каспаз 1, 3 и рекрутировании белка Bax при апоптозе, индуцированном TNF-a, интерферонами 1 и 2, активацией Fas и повреждениями ДНК [77,78]. Помимо регуляции апоптоза PML контролирует также размножение и, вероятно, дифференцировку миелоидных предшественников. Так, показано, что трансактивация p21WAF1/CIP1, ответственная за остановку клеточного цикла под воздействием ретиноевой кислоты, опосредуется именно PML [79]. Таким образом, экспрессия химерного белка PML/RAR-a, вызывающая инактивацию нормальной функции белка PML, как и перестройка BCR/ABL, ведет одновременно и к изменениям регуляции клеточного цикла, и к частичному блокированию индукции апоптоза (следует заметить, что в отличие от BCR/ABL перестройка PML/RAR-a вызывает также и блок дифференцировки; см. раздел 8). В результате многонаправленного характера действия гибридных молекул появляются клетки с повышенным пролиферативным потенциалом и одновременно с устойчивостью к негативным регуляторным сигналам и/или неблагоприятным условиям окружающей среды. Предполагается, что такие изменения могут быть уже достаточными для развития по крайней мере некоторых форм гемобластозов. И, действительно, перестройки BCR/ABL или PML/RAR-a часто являются единственными генетическими изменениями, обнаруживаемыми соответственно при хроническом миелоидном и остром промиелоцитарном лейкозах [3,12].
Однако для развития злокачественных форм солидных опухолей (раков, сарком и др.), безусловно, требуются и другие изменения, в первую очередь обусловливающие нарушения взаимодействия клеток со своими соседями и внеклеточным матриксом, и, в частности, потерю ими зависимости от субстрата и контактного торможения размножения; повышенную локомоторную активность, ответственную за инвазию в окружающие ткани и др.; способность неопластических клеток стимулировать прорастание сосудов (неоангиогенез) в ткани опухоли для обеспечения ее питания и т.д. В связи с этим неудивительно, что в клетках солидных опухолей число выявляемых мутаций и других генетических изменений, как правило, значительно выше, чем в клетках лейкозов. Число генетических перестроек нередко достигает в них нескольких десятков. Ясно, что исходя из обычного темпа мутирования, характерного для нормальных клеток, нельзя объяснить появления в одной клетке такого количества генетических нарушений. Поэтому, прежде чем перейти к анализу роли протоонкогенов и опухолевых супрессоров в регуляции морфогенетических реакций клетки и неоангиогенеза, рассмотрим механизмы возникновения генетической нестабильности - еще одного важнейшего признака неопластической клетки.