ПАТОГЕНЕЗ ЗРЕЛОКЛЕТОЧНЫХ ЛИМФАТИЧЕСКИХ ОПУХОЛЕЙ
Е.А. Никитин
Гематологический Научный Центр РАМН, Москва
Лимфомогенез складывается из цепи событий, в которой можно выделить несколько этапов: предрасполагающие факторы, первичные онкогенные события и вторичные онкогенные события. Предрасполагающими факторами могут быть хроническая антигенная стимуляция, врожденные или приобретенные иммунодефицитные состояния, вирусные инфекции. Первичным онкогенным событием, запускающим развитие опухоли, считают генетическое нарушение, выявляемое в большинстве случаев лимфом данного типа, начиная с самых ранних стадий. Примерами первичных онкогенных событий являются транслокации t(11;14) при лимфоме из клеток мантийной зоны, t(14;18) при фолликулярной лимфоме. Ко вторичным онкогенным событиям относят генетические нарушения, появляющиеся в ходе прогрессии опухоли и отягчающие ее течение. Вторичные генетические нарушения при лимфомах сравнительно однообразны по последствиям: в большинстве случаев повреждаются гены, регулирующие клеточный цикл, что усиливает пролиферацию клеток.
В данной работе представлен обзор первичных онкогенных событий и их последствий при В-клеточных зрелоклеточных лимфатических опухолях. Под зрелоклеточными лимфомами мы понимаем опухоли, возникающие из клеток периферических лимфоидных органов. Таким образом, к зрелоклеточным относится большая группа опухолей, кроме лимфобластных лимфом и лейкозов из ранних предшественников.
Особенности онкогенеза лимфом. Механизмы транслокаций.
Генетические нарушения при лимфомах весьма специфичны. Общая нестабильность генома, проявляющаяся в накоплении многочисленных хромосомных аберраций и характерная для многих типов эпителиальных опухолей, лимфатическим опухолям не свойственна. Нестабильность микросателлитных повторов, связанная с дефектами генов репарации, при лимфомах наблюдается редко. Как и при большинстве опухолей человека, генетические повреждения при лимфомах можно подразделить на две крупные категории: активация протоонкогенов и инактивация генов-супрессоров опухолевого роста. Инактивация генов, подавляющих опухолевый рост, при лимфомах происходит посредством тех же механизмов, что и при других опухолях у человека.
Ведущим механизмом активации протоонкогенов при лимфатических опухолях являются хромосомные транслокации. Большинство транслокаций при лимфомах имеет общие черты. На одной из партнерских хромосом, близко к участку рекомбинации располагается протоонкоген, который в типичном случае не изменен структурно, нарушена только регуляция его экспрессии. Такой вариант транслокации можно противопоставить транслокациям при острых лейкозах, которые обычно приводят к слиянию двух генов и образованию химерного продукта с новыми онкогенными свойствами. Оба гена-участника структурно изменены. При лимфомах протоонкоген чаще всего перемещается в область локусов генов иммуноглобулинов и попадает под влияние гетерологичных элементов, регулирующих экспрессию генов на партнерской хромосоме. Это приводит к постоянной экспрессии протоонкогена, независимой от обычных стимулов (тогда как в норме его экспрессия происходит только в ответ на эти стимулы), или к неспецифическому усилению экспрессии протоонкогена (тогда как в норме его экспрессия очень слабая). Значительно реже активация протоонкогенов при лимфомах происходит по другим механизмам, не связанным с образованием транслокаций.
В-лимфоциты отличаются от других клеток организма тем, что в процессе созревания их геном подвергается большому числу соматических перестроек ДНК. Обилие перестроек генома создает основу для онкогенных нарушений. Эти онкогенные нарушения происходят постоянно, поскольку хромосомные транслокации с участием локусов генов иммуноглобулинов с некоторой частотой выявляются в лимфоцитах здоровых людей. Так, транслокации MYC/IgH выявляются у 2% здоровых лиц, а транслокация BCL2/IgH - у 50-60% здоровых лиц, причем частота возрастает с возрастом.
Транслокации могут возникать в результате ошибок V(D)J-рекомбинации, ошибок сдвига изотипа. К V(D)J-зависимым транслокациям относятся t(11;14) при лимфоме из клеток мантии, t(14;18) при центрофолликулярной лимфоме (354), t(1;14) с участием гена bcl-9 при лимфобластной лимфоме, t(1;14) с участием гена bcl-10 при MALT-лимфомах. Во время сдвига изотипа возникает один из вариантов транслокации t(8;14), характерный для спорадической лимфомы Беркитта, t(9;14), характерный для лимфоплазмоцитоидной лимфомы. Наконец, транслокации могут появляться в результате ошибок соматической гипермутации. Например, в это время возникает вариант транслокации t(8;14), характерный для эндемичной лимфомы Беркитта. Кроме того, соматическая гипермутация может затрагивать и повреждать гены, расположенные в других локусах – BCL-6, CD95, PAX-5, MYC, Rho-TTF и PIM-1.
Таким образом, основной особенностью лимфомогенеза являются транслокации, являющиеся следствием многочисленных перестроек ДНК, происходящих в В-лимфоцитах в норме. Транслокации возникают случайно, и в большинстве случаев ни к чему не приводят: клетки-носительницы транслокаций просто погибают. Опухоль возникает, если транслокация «уместна», то есть оказывается (случайным образом) в определенной субпопуляции лимфоцитов на определенной стадии развития.
Данные о популяциях лимфоцитов традиционно базировались на иммунофенотипических исследованиях. Последнее десятилетие было ознаменовано интенсивными молекулярными исследованиями состояния генов иммуноглобулинов в лимфоцитах и клетках лимфом. На каждом этапе созревания лимфоцитов состояние генов иммуноглобулинов в них различно, поэтому эти исследования помогли существенно прояснить представления о субпопуляциях В-лимфоцитов у человека и понять гистогенез лимфом. Систематический анализ соматической гипермутации позволил подразделить все лимфатические опухоли на три группы: префолликулярные, фолликулярные и постфолликулярные (табл. 1).
Таблица 1.
Распределение лимфатических опухолей по отношению к соматической гипермутации.
| Из пре-фолликулярных клеток | Из клеток центра фолликула | Из пост-фолликулярных клеток | |
|---|---|---|---|
| Лимфома из клеток мантийной зоны | + | ≈1/4 случаев | |
| В-клеточный хронический лимфолейкоз | ≈50% | ? | ≈50% |
| Фолликулярная лимфома | + | ||
| Лимфома Беркитта | + | ||
| В-крупноклеточная лимфома | + | Часть случаев | |
| Макроглобулинемия Вальденстрема | Часть случаев | + | |
| Волосатоклеточный лейкоз | Часть случаев | + | |
| В-клеточный пролимфоцитарный лейкоз | + | ||
| Группа лимфом маргинальной зоны | Часть случаев | + | |
| Миелома | + |
Субпопуляции лимфоцитов.
Современная упрощенная схема дифференцировки В-лимфоцитов представлена на рис. 1.
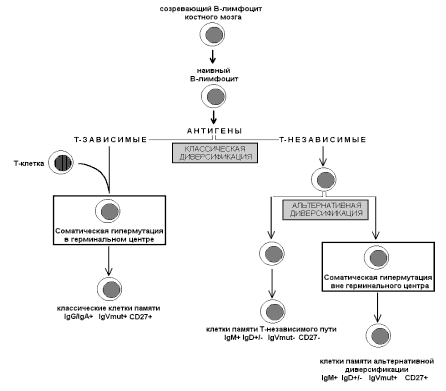
Рис. 1. Схема дифференцировки В-лимфоцитов.
В-клеточный лимфопоэз начинается в костном мозге, где в ранних предшественниках В-лимфоцитов происходят перестройки ДНК, в результате чего в составе генома каждого из них формируется участок, кодирующий вариабельный регион локуса иммуноглобулинов. В случае тяжелой цепи вариабельный регион формируется при участии VH-, DH- и JH-сегментов. Вариабельный участок легкой цепи каппа или лямбда кодируется VL- и JL-сегментами, без участия D-сегментов. В-клетки, в которых успешно завершены все перестройки, покидают костный мозг, и на этом заканчивается антиген-независимый этап созревания. Наивные В-клетки экспрессируют поверхностные иммуноглобулины классов IgM и IgD, не имеют признаков соматической гипермутации, не экспрессируют CD27.
Если в течение нескольких дней после выхода из костного мозга зрелая наивная В-клетка не активируется, в ней запускается программа апоптоза, и она погибает. Продолжение жизни В-лимфоцита возможно после активации, которая происходит при связывании с антигеном. На этом этапе происходит первая (классическая) диверсификация лимфоцитов. Часть из них реагирует с Т-зависимыми антигенами, то есть антигенами, иммунный ответ против которых требует участия Т-хелперов. В-клетки Т-зависимого пути связывают антиген, активируются и мигрируют в фолликулы периферических лимфоидных органов, где при участии Т-клеток в специфическом микроокружении герминальных центров (ГЦ) происходит их размножение, соматическая гипермутация и созревание аффинности антител В-клеток, селектированных антигеном.
Во время соматической гипермутации на небольшом по протяженности участке (1500–2000 нуклеотидов) в районе перестроенного V-(D)-J-сегмента возникает множество мутаций. Мутационный процесс не затрагивает остальной части генома. Часть мутаций приводит к аминокислотным заменам (R-мутации, replacement) и меняет белковую последовательность вариабельного региона антитела. Случайность возникновения R-мутаций, в большинстве случаев, приводит к утрате аффинности к тому антигену, который данная В-клетка связала исходно. В таком случае В-клетка погибает по механизму апоптоза. В небольшом числе клеток мутации приводят к повышению аффинности антитела к антигену. Такие В-клетки преодолевают постоянно генерируемый в ГЦ сигнал к апоптозу и продолжают делиться. В части В-клеток герминального центра происходит сдвиг изотипа (IgM/D–IgG/A), изменяющий функциональные свойства антител. Классические клетки, прошедшие стадию центра фолликула характеризуются признаками прошедшей соматической гипермутации, экспрессией IgG/A, экспрессией CD27.
Другие наивные В-клетки реагируют с Т-независимыми антигенами, к которым относятся преимущественно липополисахариды бактерий (небелковые антигены). Причины комиттирования в Т-зависимую или Т-независимую линии, по-видимому, связаны со специфичностью антител на поверхности наивных лимфоцитов. Если антитела данной клетки обладают большей аффинностью к липополисахаридам бактерий, она запускается в Т-независимый путь дифференцировки. Лимфоциты, отвечающие на Т-независимые антигены, не проходят стадию центра фолликула – самую длительную фазу активации. Благодаря этому они значительно быстрее вырабатывают антитела: высокий титр антител против Т-независимых антигенов обнаруживается уже к концу вторых суток. В-клетки Т-независимого пути являются частью защиты от бактериемии и сепсиса, поэтому скорость иммунного ответа принципиальна. Иммунный ответ против Т-независимых антигенов происходит преимущественно в селезенке. Через несколько месяцев после удаления селезенки В-клетки, отвечающие на Т-независимые антигены, практически исчезают. При врожденной асплении они не обнаруживаются совсем. Классические клетки маргинальной зоны селезенки экспрессируют только IgM и не экспрессируют IgD/IgG/IgA (IgM only cells). Они также экспрессируют CD27.
У больных с гипер-IgM синдромом, обусловленным врожденным дефектом молекулы CD40, созревание В-лимфоцитов заблокировано на стадии герминального центра. В периферических лимфоидных органах этих пациентов герминальных центров нет. В-лимфоциты не подвергаются сдвигу изотипа, поэтому у них отсутствуют клетки IgG+ IgA+, выявляются только IgM+/IgD± клетки. Тем не менее, в части лимфоцитов этих больных выявляются признаки соматической гипермутации. Эти данные легли в основу представлений о второй или альтернативной диверсификации лимфоцитов. После контакта с антигеном лимфоциты Т-независимого пути подвергаются второй диверсификации, поскольку часть из них не содержит мутаций вариабельного региона, другие – содержат. Последняя популяция приобретает мутации вариабельного региона вне центров фолликулов. Поэтому в них не бывает сдвига изотипа; они характеризуются фенотипом IgM+/IgD±. Они тоже экспрессируют CD27.
Лимфомы из клеток центра фолликула и ранних постфолликулярных стадий.
К лимфомам из клеток центра фолликула относятся центрофолликулярная лимфома (ФЛ), В-крупноклеточная лимфома и лимфома Беркитта. Все они имеют признаки не выключенной соматической гипермутации и нередко характеризуются сдвигом изотипа.
Центрофолликулярная лимфома наследует от своего нормального аналога, центроцита, особый профиль экспрессии поверхностных маркеров и тропность к герминальным центрам. В большинстве случаев ФЛ имеет фолликулярный тип роста в лимфатических узлах. Как и нормальные центроциты, клетки ФЛ активно рециркулируют между разными фолликулами и даже разными лимфоузлами, но всякий раз оказываются преимущественно в одной зоне – герминальных центрах. ФЛ наследует низкую пролиферативную активность, поскольку центроциты практически не делятся. Наоборот, они запрограммированы на гибель, которой можно избежать, только получив мощный сигнал через В-клеточный рецептор, свидетельствующий о том, что данная клетка обладает высокоаффинным антителом. Клетки фолликулярной лимфомы избегают гибели по другому механизму - в результате транслокации t(14;18), приводящей не столько к высокой экспрессии BCL-2, сколько к ее постоянству и независимости от обычных стимулов. Другие генетические повреждения обычно ассоциируются с плохим прогнозом и трансформацией в крупноклеточную лимфому.
Ген BCL-2 кодирует интегральный мембранный белок, который локализуется на митохондриях, на гладком эндоплазматическом ретикулуме и на перинуклеарной мембране. BCL-2 является членом BCL-2-семейства регуляторов апоптоза, в которое входит множество других белков. В клетке BCL-2 существует в виде высокомолекулярных комплексов, состоящих из молекул BCL-2 и BAX. Соотношение BCL-2/BAX определяет функциональную активность BCL-2. Если BAX находится в избытке, преобладают гомодимеры BAX:BAX, и клетка более склонна к апоптозу. Наоборот, если возрастает количество BCL-2, то преобладают гетеродимеры BCL-2:BAX, и клетка более устойчива к апоптозным стимулам. Противоапоптозное действие BCL-2 осуществляется по нескольким механизмам. Этот белок регулирует проницаемость мембран митохондрий и поступление в цитоплазму цитохрома С, который связывается с белком Apaf и вызывает активацию каспаз. BCL-2 запускает активацию антиоксидантного пути, предотвращающего высвобождение свободных радикалов, а также регулирует выход кальция из эндоплазматического ретикулума.
Роль гиперэкспрессии BCL-2 в развитии фолликулярной лимфомы носит сложный характер. Транслокация происходит рано, на уровне пре-В-клеток, но не препятствует дальнейшему созреванию В-клетки (гены тяжелой цепи иммуноглобулинов продуктивно перестроены на втором аллеле). Клетки-носительницы транслокации успешно проходят антиген-независимый этап и дозревают до стадии наивных В-клеток IgM/D+. Как и другие наивные клетки, многие из них погибают в течение нескольких дней, поскольку не контактируют с антигеном. Если же клетка носительница транслокации t(14;18) контактирует с антигеном, она активируется и делится. Потомки этой клетки устойчивы к апоптозу, и их пролиферация не может быть завершена обычными механизмами ограничения иммунного ответа. Размноженный клон длительно персистирует. Со временем в нем накапливаются дополнительные генетические повреждения, что приводит появлению лимфомы.
Транслокация t(14;18) также выявляется в 30-40% случаев В-крупноклеточной лимфомы, развившихся de novo, когда нет анамнестических указаний на предшествующую зрелоклеточную фолликулярную лимфому, которая подверглась трансформации.
В-крупноклеточная лимфома (В-ККЛ) гетерогенна по морфологии, клинической картине, ответу на лечение, что частично объясняется разными вариантами возникновения этой опухоли: она может возникать de novo или развиваться на фоне зрелоклеточной лимфомы в результате трансформации. Гетерогенность В-ККЛ отчасти объяснена благодаря анализу профиля экспрессии генов при этой болезни. При сравнении профилей экспрессии генов выборки случаев В-ККЛ и наивных В-клеток, активированных В-клеток, В-клеток центра фолликула и, наконец, В-клеток памяти, оказалось, что В-ККЛ подразделяется на два варианта. Один несет «роспись» центрофолликулярных клеток, другой – активированных клеток постфолликулярных стадий. Один вариант получил название В-ККЛ из центрофолликулярных клеток, другой – В-ККЛ из активированных клеток. Первый вариант поддается терапии в 75%, второй - только в 25% случаев.Состояние генов иммуноглобулинов при этих вариантах В-ККЛ также различно. При В-ККЛ из центрофолликулярных клеток механизм соматической гипермутации не выключен. При В-ККЛ из активированных клеток постгерминального уровня внутриклоновой гетерогенности нет, то есть соматическая гипермутация завершена.
Самой частой аномалией при В-ККЛ являются различные нарушения района 3q27, содержащего ген BCL-6. Белок BCL-6 выполняет регулирующую функцию и подавляет транскрипцию генов, содержащих соответствующие регуляторные последовательности. У человека высокий уровень белка BCL-6 обнаруживается исключительно в центрофолликулярных В-клетках - как в центробластах, так и в центроцитах и не выявляется в клетках пре- и постфолликулярных стадий. В отсутствие BCL-6 (у нокаутированных по BCL-6 мышей) герминальные центры не образуются. Таким образом, BCL-6 - ключевой регулятор формирования и нормальной функции герминальных центров, причем прекращение экспрессии BCL-6 необходимо для дальнейшей дифференцировки В-клеток.
Локус 3q27 не имеет постоянной хромосомы-партнера, но все транслокации с его участием характеризуются одной особенностью: кодирующая область BCL-6 всегда остается интактной; но в результате транслокации происходит замещение промотора, то есть последовательности, регулирующей экспрессию BCL-6. Поскольку нет постоянной хромосомы партнера, промоторы разные, но все они обладают одной особенностью: их активность не прекращается после того, как В-клетка проходит стадию герминального центра. В результате транслокаций экспрессия BCL-6 не может прекратиться, и это вызывает блок дальнейшей дифференцировки В-клеток в плазматические. Таким образом, клетки В-ККЛ «заморожены» на уровне герминального центра. Блок дифференцировки на стадии герминального центра особенно опасен, поскольку программа клеток герминального центра подразумевает быстрое деление.
В норме BCL-6 подавляет гены, участвующие в В-клеточной активации, воспалении и терминальной дифференцировке лимфоцитов. Одной из основных мишеней BCL-6 является белок BLIMP1 (B-Lymphocyte-Induced Maturation Protein 1), служащий ключевым регулятором терминальной дифференцировки в плазматические клетки. BLIMP1 - тоже транскрипционный репрессор, который подавляет всю программу В-клеточной линии и, таким образом, блокирует передачу сигнала через В-клеточный рецептор, сдвиг изотипа, соматическую гипермутацию и другие В-клеточные функции. Таким образом, BLIMP1 вызывает глобальные изменения, в результате чего клетки терминальной дифференцировки, то есть плазматические, очень мало напоминают по профилю экспрессии зрелые В-клетки.
В ходе нормальной дифференцировки В-клеток, BLIMP1 и BCL-6 образуют регуляторную петлю с отрицательной обратной связью: оба гена подавляют экспрессию друг друга. В герминальном центре большинство В-клеток экспрессируют BCL-6 и не экспрессируют BLIMP1. Меньшая часть клеток, возможно дифференцирующаяся в плазматические, наоборот, экспрессирует BLIMP1 и не экспрессирует BCL-6.
В физиологических условиях лимфоциты герминального центра получают мощные активационные сигналы от двух поверхностных рецепторов - CD40 (Т-клетки) и В-клеточного рецептора (антиген). Оба сигнала сильно активируют путь NF-kB, который опосредованно через IRF4 приводит к резкому падению уровня BCL-6. В результате, в клетке преобладает экспрессия BLIMP1, и запускается терминальная плазматическая дифференцировка. Таким образом, если клетка получает мощную стимуляцию от антигена и Т-клеток, она дифференцируется в плазматическую. Транслокация с участием гена BCL-6 приводит к тому, что его экспрессия больше не зависит от сигналов, поступающих через В-клеточный рецептор и CD40. В итоге происходит блок дифференцировки в плазмоциты, «замораживание» клеток-носительниц транслокаций на стадии герминального центра, что содействует бесконтрольной В-клеточной пролиферации. Длительное персистирование клетки на стадии герминального центра может приводить к появлению вторичных онкогенных нарушений, возникающих в результате ошибок соматической гипермутации и сдвига изотипа, что приводит к развитию лимфомы.
Лимфома Беркитта (ЛБ) наследует от своего нормального аналога, центробласта, способность к очень интенсивному делению. Как уже отмечалось, клетки центра фолликула совершают 1 деление за 6-7 часов. Этим объясняется, с одной стороны, крайне агрессивное течение этой опухоли, а с другой стороны, - высокая курабельность при адекватной интенсивной полихимиотерапии. Основным нарушением при лимфоме Беркитта являются транслокации с участием локусов генов Ig и 8q24, что приводит к одному нарушению регуляции экспрессии протоонкогена MYC. Белок MYC представляет собой повсеместно экспрессируемый ядерный фосфопротеин bHLH-LZIP, который работает как транскрипционный фактор, регулирующий клеточную пролиферацию, дифференцировку и апоптоз. Однако основным последствием гиперэкспрессии MYC является мощное усиление пролиферации клеток.
Уход от терминальной дифференцировки - признак лимфоплазмоцитоидной лимфомы. В 50% случаев этой опухоли выявляется транслокация t(9;14)(q13;q32) с участием локуса иммуноглобулинов и гена PAX-5. PAX-5 - транскрипционный фактор, который активирует различные гены В-клеток и необходим для комиттирования ранних гемопоэтических предшественников в В-клеточную линию. PAX-5 может подавлять экспрессию другого транскрипционного фактора XBP1, которая необходима для плазматической дифференцировки. В норме BLIMP1 непосредственно угнетает транскрипцию PAX5. В результате транслокации, экспрессия PAX-5 утрачивает зависимость от BLIMР1. PAX5 гиперэкспрессирован и блокирует плазматическую дифференцировку. Это определяет клиническую картину лимфоплазмоцитоидной лимфомы: опухолевый клон «заморожен» на стадии перехода «герминальная клетка - плазматическая клетка». В недавнем исследовании S. Sahota и соавт. показали, что в типичных случаях лимфоплазмоцитоидной лимфомы всегда имеются признаки соматической гипермутации, но нет сдвига изотипа (что подтверждается секрецией IgM). То есть, речь идет о стадии, когда соматическая гипермутация запущена, но сдвига изотипа еще нет.
Клетка происхождения волосатоклеточного лейкоза (ВКЛ) до сих пор не установлена. Гены вариабельного региона иммуноглобулинов при ВКЛ в большинстве случаев содержат мутации, хотя количество мутаций обычно небольшое: степень гомологии герминальным генам IgV при ВКЛ колеблется в пределах 1,4-6,5% с небольшим уровнем внутриклоновой гетерогенности. Необычность ВКЛ по сравнению с другими лимфомами состоит в том, что клетки этой опухоли нередко экспрессируют несколько изотипов иммуноглобулинов. Показано, что одна (!) клетка ВКЛ может экспрессировать одновременно несколько изотипов Ig, соединяющихся с одним и тем же вариабельным регионом. Чаще всего, наряду с IgM и IgD, клетки ВКЛ экспрессируют IgG3. Реже выявляются IgG1, IgG2, IgA1, и IgA2. Это показывает, что механизм сдвига изотипа в клетках опухолевого клона не выключен, а клетки ВКЛ «заморожены» на стадии сдвига изотипа, когда процессинг РНК предшествует геномной делеции, которая в норме происходит при сдвиге изотипа (см. выше). Таким образом, состояние генов иммуноглобулинов указывает на то, что ВКЛ берет свое начало из клеток центра фолликула, в которых только запущены механизмы сдвига изотипа и соматической гипермутации. При этом соматическая гипермутация еще не достигла своего пика, и сдвиг изотипа не завершен. Однако «морфологический портрет» и клиническое течение этой опухоли существенно отличаются от лимфом центра фолликула. Клетки ВКЛ характеризуется выраженной, гротескной активацией. Под активацией лимфоцита понимают комплексные изменения поверхностных рецепторов, цитоскелета, а также миграцию в определенные области, которые возникают после контакта с антигеном. Клетки ВКЛ имеют специфическую форму, экспрессируют поверхностные молекулы, ассоциированные с активацией (CD22, CD25, CD72, CD40L, CD11c, CD103), в том числе молекулы адгезии, молекулы входящие в иммунологический синапс, и другие. Генетические нарушения при ВКЛ слабо изучены. Состояние активации может быть связано с каким-то онкогенным событием или наследоваться от нормального аналога клетки ВКЛ.
Лимфомы из клеток постфолликулярных стадий и клеток памяти.
К этой категории относится обширная группа лимфом из клеток маргинальной зоны (МЗ), часть случаев хронического лимфолейкоза, возможно, часть случаев лимфомы из клеток мантийной зоны. Все эти опухоли имеют признаки завершенной соматической гипермутации. Сдвиг изотипа встречается только при некоторых из них. Это становится понятным, если учесть, что классические клетки маргинальной зоны характеризуются сильной экспрессией поверхностных IgM и не экспрессируют IgD, IgG, IgA. На наш взгляд, этой группе лимфом свойственны три особенности: выраженный тканевой тропизм, связь с антигенной стимуляцией и сравнительно благоприятный прогноз. Первый феномен объясним: клетки памяти «запоминают» не только антиген, но и место встречи с ним. Экстранодальные лимфомы маргинальной зоны длительно «остаются» на локальных стадиях. Так, например, поражение костного мозга при экстранодальных лимфомах МЗ выявляется в 0-15% случаев. Связь с антигенной стимуляцией ярко демонстрируется развитием мальтомы желудка на фоне персистирующей инфекции Helicobacter Pylori, мальтомы щитовидной железы на фоне тиреоидита Хашимото, мальтомы слюнной железы на фоне болезни/синдрома Шегрена. Наконец, относительно благоприятный по сравнению с другими лимфатическими опухолями прогноз может быть связан со значительной продолжительностью жизни клеток памяти и медленным обновлением. Иммунитет против некоторых инфекций сохраняется пожизненно, и это объясняется наличием клеток памяти, совершающих редкие неравные деления в которых одна клетка запускается в плазматическую дифференцировку, а другая остается клеткой памяти.
В большинстве случаев лимфомы маргинальной зоны селезенки VH-гены содержат мутации. Поэтому эта опухоль возникает либо из постфолликулярных клеток, либо из клеток, подвергшихся альтернативной диверсификации Т-независимого пути. Частая моноклональная секреция IgG говорит в пользу первой версии. Генетика лимфомы маргинальной зоны селезенки (МЗС) слабо изучена. В связи с частым (до 40%) обнаружением при лимфоме МЗС делеции 7q22, предполагается, что в ее патогенезе может иметь значение ген CDK6 (циклин-зависимая киназа 6).
Экстранодальные лимфомы маргинальной зоны - отдельная нозологическая группа, возникающая из лимфоидной ткани, ассоциированной со слизистыми (MALT). Эти лимфомы характеризуются совершенно особенными патогенетическими, гистологическими и клиническими признаками. MALT-лимфомы (или мальтомы) могут возникать практически в любом органе, однако, чаще они развиваются в желудке, слюнных железах и щитовидной железе, то есть в органах, в которых лимфоидная ткань появляется после хронического персистирующего воспаления. Это может происходить в ответ на инфекцию (геликобактерный гастрит) или вследствие персистирующего аутоиммунного процесса (миоэпителиальный сиалоаденит или тиреоидит Хашимото). Аномальный клон возникает на фоне персистирующей реактивной пролиферации. Со временем он замещает нормальную В-клеточную популяцию и приводит к развитию MALT-лимфомы.
В 30-50% случаев зрелоклеточных MALT-лимфом выявляется транслокация t(11;18)(q21;q21). Транслокация приводит к образованию и экспрессии химерного сливного продукта генов API2-MLT. API2 (Apoptosis Inhibitor-2) сильно экспрессируется в лимфоцитах селезенки и тимуса и может блокировать апоптоз как по внутреннему, так и по внешнему путям. MLT (MALT Lymphoma Translocation) играет важную роль в субклеточной локализации и стабильности химерного продукта. В норме API2 локализуется как в цитоплазме, так и в ядре, а MLT локализуется только в цитоплазме. По отдельности API2 и MLT быстро разрушаются протеасомами. Химерный продукт API2-MLT локализуется только в цитоплазме и длительно остается стабильным. Таким образом, транслокация приводит к появлению стабильного химерного белка, оказывающего антиапоптозное действие. MALT-лимфомы характеризуются низкой пролиферативной фракцией, поэтому основной патогенетический механизм этих опухолей - дефект апоптоза.
Хронический лимфолейкоз (ХЛЛ) с мутациями вариабельного региона (M-CLL). Популяции В-лимфоцитов памяти, которая точнее всего соответствовала бы варианту M-CLL, в норме пока не обнаружено. Поскольку В-ХЛЛ, как правило, экспрессирует IgM/D, редко – другие изотипы и имеет признаки завершенной соматической гипермутации, предшественники его клеток не принадлежат к классическому пути. Возможно, вариант M-CLL берет свое начало из клеток альтернативной диверсификации. Однако эти клетки не экспрессируют CD5.
Варианту M-CLL свойственна очень медленная прогрессия. По-видимому, он начинается с моноклонального В-клеточного лимфоцитоза (МВКЛ) или CLUS – хронического лимфоцитоза неясного значения, который выявляется у 3,5% лиц старше 40 лет. Связь между МВКЛ и ХЛЛ доказывается одинаковым иммунофенотипом (коэкспрессия CD19/CD5/CD23, слабая экспрессия CD20/CD79b/CD22/sIg, отсутствие CD38 и один тип легкой цепи Ig), наличием соматической гипермутации и общностью цитогенетических нарушений – преобладает del13q. МВКЛ отличается от В-ХЛЛ отсутствием существенного лимфоцитоза в обычном анализе крови (<5000 клеток в мкл) и обнаруживается по иммунофенотипу. Прогрессия в В-ХЛЛ нередко характеризуется бифазной кинетикой, что свидетельствует о появлении вторичного онкогенного события. Оно происходит редко: частота развития В-ХЛЛ в общей популяции увеличивается с возрастом и у лиц старше 70 достигает 20 на 100.000 населения. Из сопоставления порядков (3 случая на 100 МБКЛ против 3–20 на 100.000 В-ХЛЛ) видно, что В-ХЛЛ возникает в 1000 раз реже МБКЛ. Иными словами, только у одного из 1000 носителей МБКЛ разовьется В-ХЛЛ. Возможно, что одним из важных аспектов патогенеза варианта M-CLL является возрастное сужение В-клеточного репертуара.
Лимфомы из клеток, не проходивших стадию соматической гипермутации.
К этой группе относится В-ХЛЛ без мутаций вариабельного региона и большинство случаев лимфомы из клеток мантийной зоны. Около 25% IgM+IgD+ B-клеток в периферической крови взрослого (или 15% от общего числа В-клеток) экспрессируют CD5, причем эти клетки не имеют мутаций вариабельного региона Ig. По своим характеристикам эта популяция наиболее близка варианту U-CLL. Предположительно, вариант В-ХЛЛ без мутаций IgV и лимфома из клеток мантийной зоны возникают из этих клеток (CD5+ IgM+IgD+).
Развитие В-ХЛЛ без мутаций вариабельного региона (U-CLL) характеризуется прогрессирующим течением, особым спектром цитогенетических нарушений. Делеция 13q как единственная аномалия встречается в 26% случаев, трисомия 12 - в 19%, делеция 11q - в 27%, делеция 17p - в 10%. Выявление повторяющихся аномалий показывает, что протоонкогены, повреждаемые в результате этих аберраций, играют какую-то роль в клетках, из которых возникает этот вариант В-ХЛЛ. Часто выявляются делеции 11q, трисомия 12 хромосомы, делеция 17p.
Наиболее вероятным кандидатом на роль тумор-супрессорного гена при В-ХЛЛ в районе 11q22.3-q23.1 является ген ATM (Ataxia Teleangiectasia Mutated). В настоящее время доказано, что ATM принимает участие в патогенезе Т-пролимфоцитарного лимфолейкоза. В нескольких исследованиях было показано, что ATM содержит мутации и не экспрессируется при В-ХЛЛ с делецией 11q. Делеции 17р13 повреждают опухолевый супрессор TP53, кодирующий белок р53, принимающий участие в индукции апоптоза при повреждении генома клетки. Трисомия 12 как генетическая аберрация, вероятно, ведет к увеличению "дозы гена". Наиболее интересен в плане возможного влияния эффекта “увеличения дозы гена” MDM2, который связывается с р53 и инактивирует его. Сверхэкспрессия MDM2 может приводить к инактивации р53 и, таким образом, к блокированию запрограммированной клеточной гибели, индуцируемой повреждениями генома.
При лимфоме из клеток мантийной зоны постоянной аномалией является транслокация t(11;14)(q13;q32), которая приводит к гомотопной дерегуляции гена ССND1, кодирующего циклин D1, член семейства циклинов D-типа. Как и другие циклины типа D, циклин D1 действует как сенсор ростовых факторов, связующий внеклеточные сигналы с механизмами клеточного цикла и способствующий переходу клеток из фазы G1 в фазу S. Циклины группы D (D1-3) связываются с Сdk4 или Сdk6. Активные комплексы циклин/Cdk фосфорилируют и инактивируют белки семейства ретинобластомы (pRb и Rb-подобные белки р105 и р130), которые являются негативными регуляторами перехода из фазы G1 в S-фазу. В покоящихся клетках pRb и его гомологи дефосфорилированы и находятся в комплексах с транскрипционными факторами E2F-DP. Внеклеточные ростовые факторы (для В-лимфоцитов ключевыми факторами являются антигены) индуцируют синтез циклинов и циклин-зависимых киназ. Комплексы циклин/CDK фосфорилируют pRb и его гомологи, что приводит к высвобождению белков E2F-DP. В свободной форме белки E2F-DP поступают в ядро и индуцируют экспрессию генов, запускающих клеточный цикл. Гиперэкспрессия циклина D1, таким образом, может приводить к постоянной активации pRb, что способствует клеточной пролиферации. Однако, в экспериментах с трансфекцией было показано, что сам по себе циклин D1 обладает слабой малигнизирующей способностью, и его искусственная гиперэкспрессия не достаточна для развития лимфомы. В норме циклин D1 связывает p27Kip1 и может изменять уровень p27Kip1 в клетке. Возможно, что основное последствие гиперэкспрессии циклина D1 состоит именно в секвестрации p27Kip1. р27Kip1 – ингибитор CDK, который играет критическую роль в регуляции перехода G1/S на стадии сверочной точки (checkpoint), предшествующей началу синтеза ДНК. p27Kip1 эффективно связывается с комплексами циклин D/CDK4 а также, в меньшей степени, с комплексами циклин E/CDK2. Связывание p27Kip1 с D/CDK4 приводит к стабилизации комплекса, но не подавляет его активность. Наоборот, связывание p27Kip1 с E/CDK2 ингибирует киназную активность, и это тормозит клеточный цикл. В норме в фазе G1 уровень циклинов D повышается и конкурирует с циклинами E за несвязанный p27Kip1. В это время киназа CAK начинает активировать CDK2. Эти два события приводят к снижению уровня p27Kip1, во-первых, потому что он связан, а во-вторых, потому, что активированная CDK2 фосфорилирует p27Kip1 и запускает его деградацию по убиквитиновому пути. Таким образом, влияние p27Kip1 устраняется, и клетка совершает фазовый переход G1-S. Из-за огромного избытка циклина D1 при лимфоме из клеток мантийной зоны весь p27Kip1 может быть связан и секвестрирован в комплексах циклин D/CDK. Комплексы E/CDK2 при этом активированы, что содействует клеточной пролиферации.
Таким образом, представления о лимфатических опухолях претерпели существенные изменения. Изменился сам принцип классифицирования лимфом. Определение каждой нозологически отдельной лимфатической опухоли сегодня зависит не столько от «внешних признаков» - морфологии и клинической картины, сколько от того, из какой субпопуляции лимфоцитов она возникла, и какие онкогенные события произошли в клетке-предшественнице этого типа лимфомы. Эти факторы очень тесно связаны между собой, поскольку каждый этап созревания лимфоцита происходит в результате работы определенных генетических программ. Изменения состояния ключевых генов, активирующих или выполняющих программу созревания лимфоцита, а также определяющих его функцию на данном этапе, могут приводить к развитию опухоли. Как и любой другой онкогенез, лимфомогенез опирается на нормальные механизмы дифференцировки, апоптоза и клеточного цикла. В справедливости этих представлений убеждает значительное сходство «биологического поведения» вариантов лимфом с их нормальными аналогами и постоянство ассоциации вариантов лимфом с определенными генетическими изменениями.